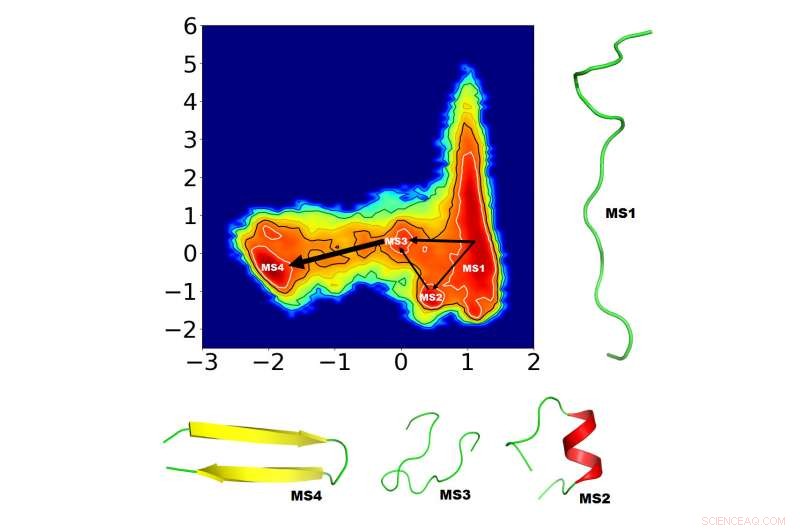
Os cientistas buscam entender melhor o dobramento de proteínas para curar doenças de dobramento incorreto, mas este processo incrivelmente complexo requer algoritmos sofisticados para identificar os mecanismos de dobramento. Biofísicos computacionais propuseram uma nova maneira de identificar os fatores mais cruciais para o enovelamento de proteínas. Eles demonstraram o curto tempo de simulação de sua abordagem em uma proteína pequena, mas intrigante, "GB1 beta-hairpin, " no Journal of Chemical Physics . Os quatro novos estados de dobramento intermediário (MS1-4) identificados pela equipe são mostrados aqui, junto com as possíveis vias de conexão. A espessura das setas de interconexão refletem a probabilidade de que o caminho ocorra. Crédito:Navjeet Ahalawat e Jagannath Mondal
Os padrões de dobramento de uma proteína os ajudam a realizar suas tarefas dedicadas. Como os verdadeiros "fazedores" da célula, mesmo uma pequena alteração na estrutura de aminoácidos de uma proteína pode causar dobramento incorreto e prejudicar a funcionalidade da proteína ou causar doenças. Por exemplo, se tau, uma proteína que ajuda a estabilizar a estrutura das células cerebrais, está mal dobrado, pode formar emaranhados tau, que são comumente vistos em pacientes com Alzheimer.
Os cientistas buscam entender melhor o dobramento de proteínas para curar doenças de dobramento incorreto, mas este processo incrivelmente complexo requer algoritmos sofisticados para identificar os mecanismos de dobramento. Biofísicos computacionais do Instituto Tata de Pesquisa Fundamental de Hyderabad (TIFR-H) propuseram uma nova maneira de identificar os fatores mais cruciais para o enovelamento de proteínas. Eles demonstraram o curto tempo de simulação de sua abordagem em uma proteína pequena, mas intrigante, "GB1 beta-hairpin, " no Journal of Chemical Physics , da AIP Publishing.
"Ao combinar um método conhecido como 'Análise de componente independente baseada na estrutura do tempo' (TICA) com simulações curtas de dinâmica molecular, encontramos quatro estados de dobramento intermediários fisicamente significativos, não observado anteriormente, e mostrou estados helicoidais que geralmente não podem ser detectados por outros métodos, "disse Navjeet Ahalawat, um autor no papel.
Cada átomo em uma proteína pode se dobrar em três dimensões, mas com milhões de átomos presentes mesmo em proteínas simples, a tarefa de compreender a combinação de dobradura coletiva torna-se complicada. Os cientistas consideraram os diferentes fatores que influenciam o enovelamento de proteínas, como ligações de hidrogênio, e combinados em descrições gerais chamadas variáveis coletivas (CVs). Contudo, com muitos fatores potenciais, os cientistas não têm uma boa maneira de encontrar currículos que descrevam apropriadamente um processo viável.
"Existem muitas maneiras pelas quais as proteínas podem ir de estados desdobrados para dobrados, então, o mais desafiador é decidir por onde começar, "Ahalawat disse. Jagannath Mondal, outro autor no papel, acrescentou que era fácil "se perder nos dados".
A equipe decidiu estudar o grampo externo saliente da proteína GB1 por causa do grande volume de trabalho existente e muitas possibilidades de dobramento potenciais já estimadas em currículos anteriores. Ahalawat e Mondal pegaram vários CVs GB1 existentes como CVs constituintes e os combinaram linearmente usando a TICA para identificar um par de CVs "otimizados". Então, eles inseriram os CVs otimizados no Modelo de Estado de Markov e identificaram quatro estados de dobramento intermediários junto com as possíveis vias de conexão.
"Nós perguntamos, quais são as características estimadas anteriormente para esta proteína em particular que pode realmente desempenhar um papel fundamental no sistema? E podemos encontrar a combinação certa de condições? ", Disse Ahalawat." Em nosso trabalho, agora podemos dizer quantitativamente se essa característica é relevante para o processo. "
"Usando simulações curtas, descobrimos o peso que você realmente precisa usar em uma combinação, e isso dá o padrão de dobramento correto para uma proteína, "Mondal acrescentou." É uma maneira realmente barata de descobrir o enovelamento de proteínas. "
Em seu método, dados de estudos anteriores são necessários para identificar CVs ideais. A equipe prevê que sua técnica pode ser usada para descobrir o mecanismo interno de dobramento de proteínas saudáveis para corrigir doenças que causam proteínas mal dobradas. Eles também desejam desenvolver ainda mais seu método de otimização de CV e aplicá-los no reconhecimento biomolecular e na descoberta de medicamentos. "No futuro, planejamos incorporar métodos não lineares, usando técnicas de aprendizado profundo baseadas em redes neurais para melhorar nosso modelo, "Ahalawat disse.