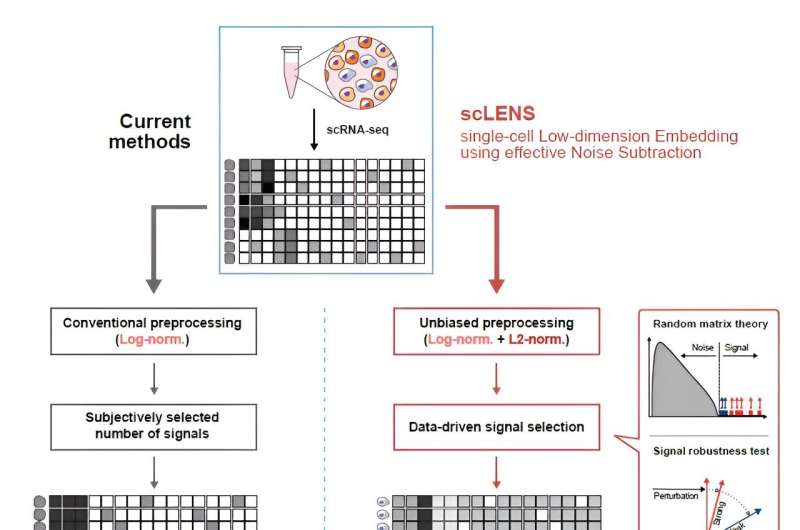
 Visão geral do scLENS (incorporação de baixa dimensão de célula única usando o Noise Subtract eficaz). (Esquerda) Os métodos atuais de redução de dimensionalidade para dados scRNA-seq envolvem etapas convencionais de pré-processamento de dados, como normalização de log, seguida pela seleção manual de sinais dos dados dimensionados. No entanto, este estudo revela que os altos níveis de esparsidade e variabilidade nos dados scRNA-seq podem levar à distorção do sinal durante o pré-processamento dos dados, comprometendo a precisão das análises downstream. (Direita) Para resolver esse problema, os pesquisadores integraram a normalização L2 no pipeline de pré-processamento convencional, mitigando efetivamente a distorção do sinal. Além disso, eles desenvolveram um novo algoritmo de detecção de sinal que elimina a necessidade de intervenção do usuário, aproveitando a filtragem de ruído baseada na teoria da matriz aleatória e os testes de robustez do sinal. Crédito:Nature Communications (2024). DOI:10.1038/s41467-024-47884-3
Visão geral do scLENS (incorporação de baixa dimensão de célula única usando o Noise Subtract eficaz). (Esquerda) Os métodos atuais de redução de dimensionalidade para dados scRNA-seq envolvem etapas convencionais de pré-processamento de dados, como normalização de log, seguida pela seleção manual de sinais dos dados dimensionados. No entanto, este estudo revela que os altos níveis de esparsidade e variabilidade nos dados scRNA-seq podem levar à distorção do sinal durante o pré-processamento dos dados, comprometendo a precisão das análises downstream. (Direita) Para resolver esse problema, os pesquisadores integraram a normalização L2 no pipeline de pré-processamento convencional, mitigando efetivamente a distorção do sinal. Além disso, eles desenvolveram um novo algoritmo de detecção de sinal que elimina a necessidade de intervenção do usuário, aproveitando a filtragem de ruído baseada na teoria da matriz aleatória e os testes de robustez do sinal. Crédito:Nature Communications (2024). DOI:10.1038/s41467-024-47884-3 Desbloquear informações biológicas de dados genômicos unicelulares complexos acaba de se tornar mais fácil e preciso, graças à inovadora ferramenta scLENS desenvolvida pelo Grupo de Matemática Biomédica do Centro IBS de Ciências Matemáticas e Computacionais, liderada pelo Investigador Chefe Kim Jae Kyoung, que também é Professor da KAIST. Isso representa um salto significativo no campo da transcriptômica unicelular.
A pesquisa foi publicada na revista Nature Communications .
A análise genômica unicelular é uma técnica avançada que mede a expressão gênica no nível celular individual, revelando alterações e interações celulares que não são observáveis com os métodos tradicionais de análise genômica. Quando aplicada a tecidos cancerosos, esta análise pode delinear a composição de diversos tipos de células dentro de um tumor, fornecendo informações sobre como o câncer progride e identificando genes-chave envolvidos durante cada estágio de progressão.
Apesar do imenso potencial da análise genômica unicelular, lidar com a vasta quantidade de dados que ela gera sempre foi um desafio. A quantidade de dados cobre a expressão de dezenas de milhares de genes em centenas a milhares de células individuais. Isto não só resulta em grandes conjuntos de dados, mas também introduz distorções relacionadas ao ruído, que surgem em parte devido às atuais limitações de medição.
O autor correspondente, Kim Jae Kyoung, destacou:"Houve um avanço notável nas tecnologias experimentais para análise de transcriptomas unicelulares na última década. No entanto, devido às limitações nos métodos de análise de dados, tem havido uma luta para utilizar plenamente dados valiosos obtidos através custos e tempo extensos."
Os pesquisadores desenvolveram vários métodos de análise ao longo dos anos para discernir sinais biológicos desse ruído. No entanto, a precisão destes métodos tem sido menos que satisfatória. Uma questão crítica é que a determinação dos limites de sinal e ruído muitas vezes depende de decisões subjetivas dos usuários.
A ferramenta scLENS recentemente desenvolvida aproveita a Teoria da Matriz Aleatória e o teste de robustez de sinal para diferenciar automaticamente sinais de ruído sem depender de informações subjetivas do usuário.
O primeiro autor, Kim Hyun, afirmou:"Anteriormente, os usuários tinham que decidir arbitrariamente o limite de sinal e ruído, o que comprometia a reprodutibilidade dos resultados da análise e introduzia a subjetividade. O scLENS elimina esse problema detectando automaticamente sinais usando apenas a estrutura inerente dos dados."
Durante o desenvolvimento do scLENS, os pesquisadores identificaram as razões fundamentais para imprecisões nos métodos de análise existentes. Eles descobriram que os métodos de pré-processamento de dados comumente usados distorcem os sinais biológicos e o ruído. A nova abordagem de pré-processamento oferecida pelo scLENS está livre de tais distorções.
Ao resolver problemas relacionados ao limite de ruído determinado pela escolha subjetiva do usuário e pela distorção do sinal no pré-processamento de dados convencional, o scLENS supera significativamente os métodos existentes em termos de precisão. Além disso, o scLENS automatiza o laborioso processo de seleção da dimensão do sinal, permitindo aos pesquisadores extrair sinais biológicos de forma conveniente e automática.
Ci Kim acrescentou:"o scLENS resolve problemas importantes na análise de dados do transcriptoma unicelular, melhorando substancialmente a precisão e a eficiência em todo o processo de análise. Este é um excelente exemplo de como as teorias matemáticas fundamentais podem impulsionar a inovação na pesquisa em ciências da vida, permitindo aos pesquisadores mais responda com rapidez e precisão a questões biológicas e descubra segredos da vida que antes estavam escondidos."