O estudo oferece uma poderosa abordagem de modelagem computacional para simulações de células
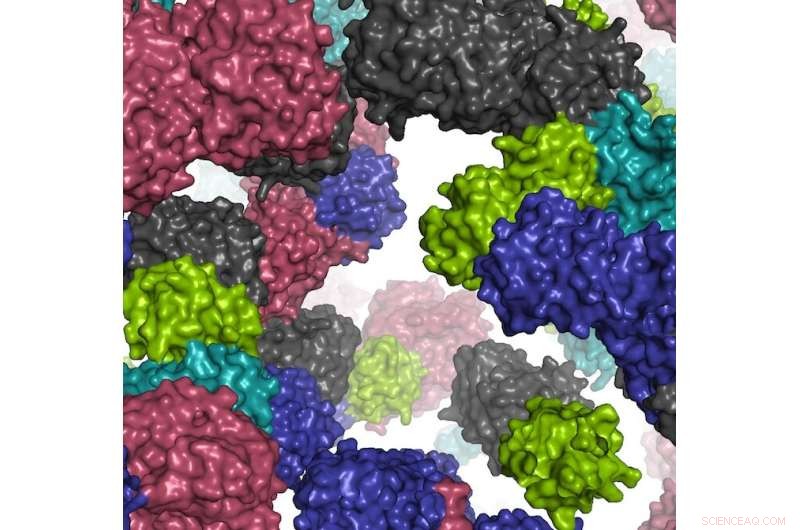
Um fragmento do ambiente celular simulado. Crédito:Ilya Vakser
Um relatório importante da Universidade do Kansas publicado esta semana na revista
Proceedings of the National Academy of Sciences propõe uma nova técnica para modelar a vida molecular com computadores.
De acordo com o principal autor Ilya Vakser, diretor do Programa de Biologia Computacional e Centro de Biologia Computacional e professor de biociências moleculares da KU, a investigação sobre modelagem computacional de processos de vida é um passo importante para criar uma simulação funcional de uma célula viva em resolução atômica. . O avanço promete novos insights sobre a biologia fundamental de uma célula, bem como um tratamento mais rápido e preciso de doenças humanas.
"É cerca de dezenas ou centenas de milhares de vezes mais rápido do que as técnicas de resolução atômica existentes", disse Vakser. “Isso oferece oportunidades sem precedentes para caracterizar mecanismos fisiológicos que agora estão muito além do alcance da modelagem computacional, obter insights sobre mecanismos celulares e usar esse conhecimento para melhorar nossa capacidade de tratar doenças”.
Até agora, um grande obstáculo para a modelagem de células via computador tem sido como abordar as proteínas e suas interações que estão no centro dos processos celulares. Até o momento, as técnicas estabelecidas para modelar interações de proteínas dependiam de "acoplamento de proteínas" ou "simulação molecular".
De acordo com os investigadores, ambas as abordagens têm vantagens e desvantagens. Embora os algoritmos de ancoragem de proteínas sejam ótimos para amostragem de coordenadas espaciais, eles não levam em conta a "coordenada de tempo" ou a dinâmica das interações de proteínas. Por outro lado, as simulações moleculares modelam bem a dinâmica, mas essas simulações são muito lentas ou de baixa resolução.
“Nosso estudo de prova de conceito une as duas metodologias de modelagem, desenvolvendo uma abordagem que pode alcançar escalas de tempo de simulação sem precedentes em resolução de todos os átomos”, escreveram os autores.
Os colaboradores de Vakser no jornal foram Sergei Grudinin, da Universidade de Grenoble Alpes, na França; Eric Deeds da Universidade da Califórnia-Los Angeles; O estudante de doutorado da KU Nathan Jenkins e Petras Kundrotas, professor assistente de pesquisa do Programa de Biologia Computacional da KU.
Depois de conceituar a melhor forma de combinar as vantagens das duas abordagens de modelagem de proteínas, a equipe desenvolveu e codificou um algoritmo para conduzir a nova simulação.
"O desafio mais difícil foi desenvolver o algoritmo que refletisse adequadamente a ideia básica simples da abordagem", disse Vakser.
Mas uma vez que eles fizeram esse avanço, eles poderiam começar a validar o novo procedimento.
"O paradigma foi fácil - um golpe de clareza", disse Vakser.
"As abordagens de simulação existentes gastam a maior parte do tempo de computação viajando em áreas de baixa probabilidade - ou de alta energia - do sistema. Todos nós sabemos onde estão essas áreas. -probabilidade, áreas de baixa energia, e pular as de baixa probabilidade estimando as taxas de transição entre os estados de alta probabilidade. O paradigma é tão antigo quanto a própria modelagem biomolecular e tem sido amplamente utilizado desde o início da era da modelagem décadas atrás."
Mas Vakser disse que até o novo artigo de sua equipe, a abordagem não havia sido aplicada à cinética das interações de proteínas no ambiente celular, o foco de seu estudo.
“Como há muito menos estados de alta probabilidade do que os de baixa probabilidade, isso nos deu um enorme ganho na velocidade de cálculo – dezenas a centenas de milhares de vezes”, disse Vakser. "Isso foi feito sem perda aparente de precisão. Pode-se argumentar que a precisão foi ganha, porque o protocolo de simulação é baseado nas técnicas de 'docking', que são projetadas especificamente para caracterizar conjuntos de proteínas."
O pesquisador da KU disse que seu método de simulação de células pode ser implantado para pesquisar a saúde humana e tratar doenças com um novo nível de precisão.
"A abordagem pode ser usada para estudar as vias moleculares subjacentes aos mecanismos da doença", disse Vakser. "Ele pode ser usado para determinar os efeitos nocivos de mutações genéticas pelos padrões alterados de associações de proteínas - mutações genéticas causam mudanças na estrutura das proteínas, que por sua vez afetam a associação de proteínas. detectar elementos críticos em padrões de associação de proteínas."
De acordo com Vakser, a nova técnica de simulação oferece muitos caminhos de pesquisa promissores para explorar no futuro.
"Entre eles estão a adaptação da abordagem de interações de proteínas com ácidos nucleicos, RNA e DNA", disse ele. "Além disso, gostaríamos de explicar a flexibilidade das formas moleculares, correlacionar com o espectro em rápido desenvolvimento de estudos experimentais do ambiente celular e aplicar o procedimento a um modelo de uma célula real - com seus componentes moleculares reais agrupados".
+ Explorar mais Ciência à beira da compreensão 'transformacional' da vida por meio de modelagem celular, dizem pesquisadores