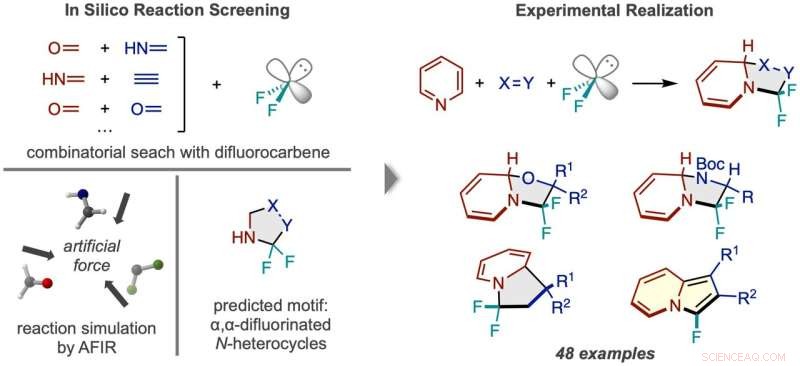
Fluxo de trabalho de descoberta de reação via triagem in silico. (Esquerda) Reações entre difluorocarbeno e vários pares de pequenas moléculas foram simuladas, prevendo um produto N-heterociclo fluorado duas vezes no carbono alfa. (Direita) A estrutura de reação bem-sucedida usando piridina e exemplos dos tipos de compostos do produto obtidos. Crédito:Síntese da Natureza (2022). DOI:10.1038/s44160-022-00128-y
As simulações de computador são mais frequentemente usadas como um guia para que os químicos possam trabalhar com mais eficiência os detalhes exatos de uma ideia de reação geral que eles têm em mente – assim como uma bússola ajuda a guiar um explorador de forma eficiente para um destino em seu mapa. No entanto, os pesquisadores do ICReDD levaram as coisas um grande passo adiante e usaram simulações para produzir a ideia geral de uma reação totalmente inimaginável, efetivamente usando cálculos para fazer o próprio mapa. Usando o princípio de design sugerido pelos resultados computacionais, a equipe atingiu o filão-mãe no laboratório, desenvolvendo com sucesso um conjunto de 48 reações que produzem compostos potencialmente úteis para o desenvolvimento de novos medicamentos.
A presença e a posição do flúor em uma molécula geralmente afetam a atividade farmacológica de uma molécula. Pesquisadores do ICReDD utilizaram cálculos químicos quânticos para descobrir uma reação que adiciona seletivamente dois átomos de flúor a uma posição de difícil acesso em um N-heterociclo – moléculas com uma estrutura de anel de carbono onde pelo menos um carbono no anel é substituído por nitrogênio . A capacidade de anexar átomos de flúor ao "carbono alfa" anteriormente difícil de acessar - o carbono imediatamente próximo ao nitrogênio na estrutura do anel - pode levar ao desenvolvimento de uma série de novas drogas.
Antes de realizar experimentos no laboratório, os pesquisadores lançaram uma ampla rede, testando computacionalmente a viabilidade de inúmeras reações de 3 componentes usando o método de reação induzida por força artificial (AFIR). Eles simularam a reação de uma molécula de difluorocarbeno, que atua na fonte de átomos de flúor, com vários pares de pequenas moléculas apresentando uma ligação dupla ou tripla. Essas simulações mostraram que várias reações de formação de anéis deveriam ser viáveis.
Os pesquisadores tentaram uma das reações promissoras sugeridas pelos resultados computacionais iniciais, mas não tiveram sucesso. Uma computação otimizada e focada mais estreitamente da energia do estado de transição da reação em questão mostrou que a molécula de difluorocarbeno reagiu mais facilmente consigo mesma do que com as moléculas iniciais desejadas, sinalizando que uma reação lateral indesejada provavelmente estava ocorrendo. This result inspired researchers to change one of the starting materials to the cyclic molecule pyridine, which they anticipated would be able to compete with the unwanted side reaction. This change resulted in the successful synthesis of the desired N-heterocyclic product with two fluorines attached at the alpha carbon position.
The reaction developed here is also significant because it breaks the aromatic system of electrons in the pyridine molecule, a transformation that is especially difficult due to the high stability of aromatic systems. Additionally, the 3-component reaction framework was applied successfully in the lab to a wide range of starting materials, resulting in many new molecules with unique alpha position fluorine substitutions. The large scope of reactivity greatly increases the potential utility of this reaction framework in new drug development.
The researchers see their streamlined screening method as a way to broaden the scope of their search and discover new horizons in chemical reaction design.
"Our study's highlight is the successful demonstration of an in silico reaction screening strategy for reaction development. The computational reaction simulation suggested less-explored three-component reactions of difluorocarbene and two unsaturated molecules, which we successfully realized in experiments," explained lead author Hiroki Hayashi.
"I think the AFIR method is a powerful tool for dictating new research directions in reaction discovery, and we plan to continue building a computation-based reaction development platform by integrating the computational and informatics techniques of ICReDD."
The study was published in
Nature Synthesis .
+ Explorar mais Hitting rewind to predict multi-step chemical reactions