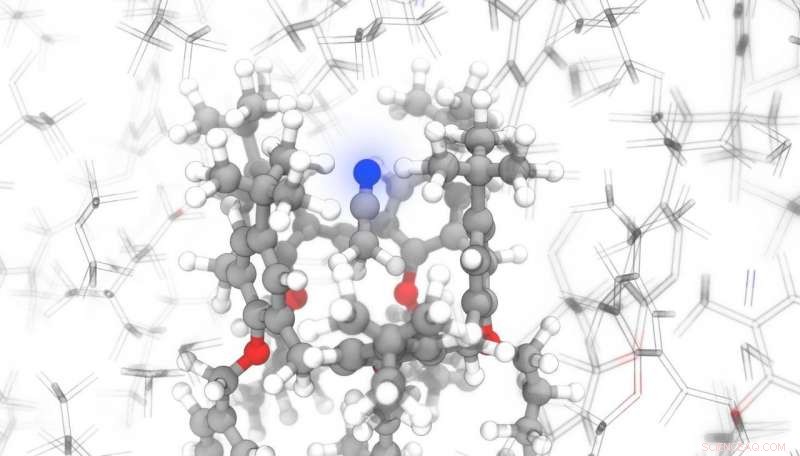
Crédito:Michele Ceriotti / EPFL
Muitos medicamentos hoje são produzidos como sólidos em pó. Mas para entender completamente como os ingredientes ativos se comportarão uma vez dentro do corpo, os cientistas precisam saber sua estrutura exata em nível atômico. Por exemplo, a forma como as moléculas são organizadas dentro de um cristal tem um impacto direto nas propriedades de um composto, como sua solubilidade. Os pesquisadores estão, portanto, trabalhando duro para desenvolver tecnologias que possam identificar facilmente as estruturas cristalinas exatas dos pós microcristalinos.
Uma equipe de cientistas da EPFL já escreveu um programa de aprendizado de máquina que pode prever, em tempo recorde, como os átomos responderão a um campo magnético aplicado. Isso pode ser combinado com a espectroscopia de ressonância magnética nuclear (NMR) para determinar a localização exata dos átomos em compostos orgânicos complexos. Isso pode ser um grande benefício para as empresas farmacêuticas, que devem monitorar cuidadosamente as estruturas de suas moléculas para atender aos requisitos de segurança do paciente. A pesquisa deles foi publicada em Nature Communications .
Velocidades alucinantes com IA
A espectroscopia de NMR é um método bem conhecido e altamente eficiente para sondar os campos magnéticos entre os átomos e determinar como os átomos vizinhos interagem uns com os outros. Contudo, a determinação total da estrutura de cristal por espectroscopia de NMR requer extremamente complicada, cálculos demorados envolvendo química quântica - quase impossível para moléculas com estruturas muito complexas.
Mas o programa desenvolvido na EPFL pode superar esses obstáculos. Os cientistas treinaram seu modelo de IA em estruturas moleculares retiradas de bancos de dados estruturais. "Mesmo para moléculas relativamente simples, este modelo tem quase 10, 000 vezes mais rápido do que os métodos existentes, e a vantagem cresce tremendamente ao considerar compostos mais complexos, "diz Michele Ceriotti, chefe do Laboratório de Ciência e Modelagem Computacional da Escola de Engenharia da EPFL e co-autor do estudo. "Para prever a assinatura de NMR de um cristal com quase 1, 600 átomos, nossa técnica - ShiftML - requer cerca de seis minutos; a mesma façanha levaria 16 anos com as técnicas convencionais. "
Este novo programa permitirá a utilização de abordagens completamente diferentes, que serão mais rápidas e permitirão o acesso a moléculas maiores. "Isso é realmente empolgante porque a aceleração maciça nos tempos de computação nos permitirá cobrir espaços conformacionais muito maiores e determinar corretamente as estruturas onde antes não era possível. Isso coloca a maioria das moléculas de drogas contemporâneas complexas ao nosso alcance, "diz Lyndon Emsley, chefe do Laboratório de Ressonância Magnética da Escola de Ciências Básicas da EPFL e coautor do estudo.
O programa agora está disponível gratuitamente online. "Qualquer pessoa pode carregar uma molécula e obter sua assinatura NMR em apenas alguns minutos, "diz Ceriotti.