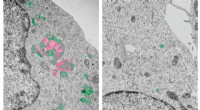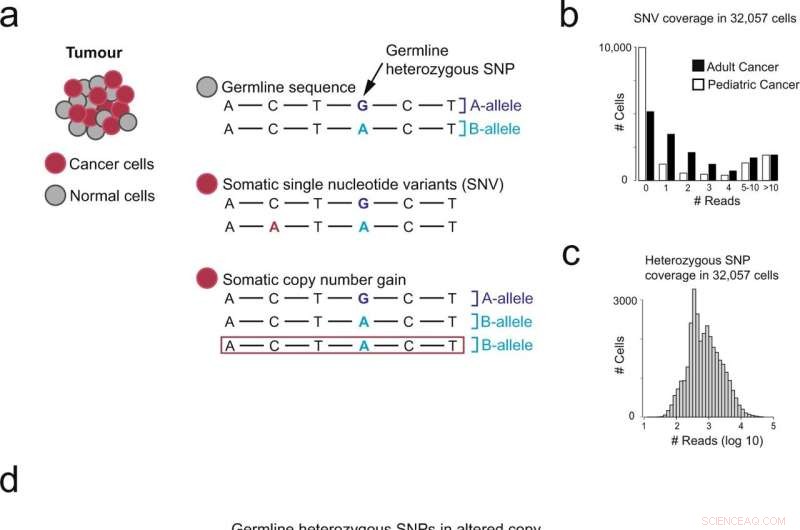
Visão geral de diferentes abordagens para identificar células derivadas de câncer. a Alterações genômicas presentes em genomas de câncer. b Número de células (eixo y) com leituras N cobrindo mutações pontuais (eixo x), separadas por carga de mutação baixa (neuroblastoma NB) e alta (carcinoma de células renais CCR). c Número de células (eixo y) com leituras N cobrindo polimorfismos de nucleotídeo único heterozigotos (SNP) (eixo x). d Visão geral do uso de mudanças alélicas representando alterações no número de cópias para detectar células cancerígenas. Crédito:Biologia da Comunicação (2022). DOI:10.1038/s42003-022-03808-9. https://www.nature.com/articles/s42003-022-03808-9
Um novo método de separação de células cancerosas de células não cancerosas foi desenvolvido por pesquisadores do Wellcome Sanger Institute, em um impulso para aqueles que trabalham para entender melhor a biologia do câncer usando o sequenciamento de mRNA de célula única.
O estudo, publicado hoje na revista
Communications Biology , melhora os métodos existentes para identificar quais células em uma amostra são cancerígenas e fornece dados cruciais sobre o microambiente dos tumores. O software está disponível abertamente para pesquisadores de todo o mundo aplicarem em seus próprios dados, aumentando a eficácia do sequenciamento de uma única célula para entender o câncer.
A análise de mRNA de célula única de células cancerígenas é uma das técnicas de ponta usadas para entender melhor a biologia do câncer. Os dados gerados podem ser usados para tentar interromper os cânceres com drogas ou descobrir como os cânceres surgem em primeiro lugar.
Um passo fundamental nesse processo é separar as células cancerígenas das não cancerosas, mas nem sempre é uma tarefa fácil. Além dos muitos tipos de câncer, também haverá diferenças moleculares entre as células cancerígenas do mesmo tipo dentro de um único tumor.
Atualmente, o melhor método de fazer isso é medir a expressão gênica média das células na amostra, com maior ou menor expressão usada para distinguir células cancerosas de células saudáveis. Mas esse método pode levar a conclusões falsas.
Neste novo estudo, pesquisadores do Instituto Wellcome Sanger realizaram sequenciamento de genoma completo e sequenciamento de mRNA de célula única em amostras coletadas pelo Great Ormond Street Hospital (GOSH).
Ao localizar desequilíbrios de alelos nesses dados, que indicam alterações no número de cópias no genoma, a equipe conseguiu identificar células cancerígenas de forma mais confiável do que com métodos anteriores. Essa abordagem será útil principalmente para validar novos tipos de células cancerígenas e entender melhor o microambiente do tecido tumoral.
"Ser capaz de saber como o transcriptoma é diferente em células com genomas aberrantes, como os encontrados em cânceres, é um conhecimento valioso e expandirá as perguntas que podemos responder usando o sequenciamento de célula única", diz o Dr. Matt Young.
O método, chamado alleleIntegrator, está disponível como um pacote de software para uso de pesquisadores de todo o mundo.
+ Explorar mais A estimativa do nível de mRNA total específico do tumor prevê os resultados do câncer