Um novo método húngaro pode ajudar na pesquisa de proteínas
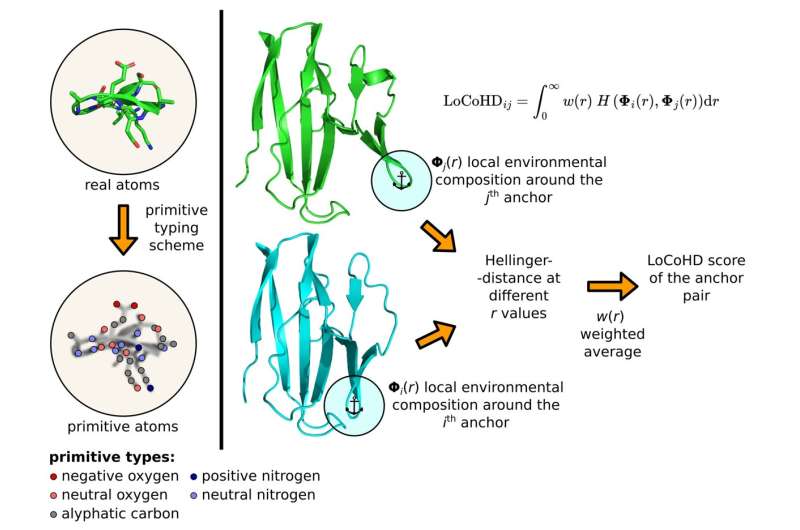 A figura mostra o processo passo a passo de cálculo da pontuação LoCoHD para um determinado par de âncoras. A resolução química é dada pelo esquema de tipagem primitiva, que é utilizado para converter a estrutura real em uma estrutura de átomos primitivos (esquerda). Então, selecionando dois átomos âncora das duas estruturas primitivas a serem comparadas (no meio, em verde e turquesa), um número entre 0 e 1 pode ser calculado usando a fórmula vista acima. Crédito:Universidade Eötvös Loránd
A figura mostra o processo passo a passo de cálculo da pontuação LoCoHD para um determinado par de âncoras. A resolução química é dada pelo esquema de tipagem primitiva, que é utilizado para converter a estrutura real em uma estrutura de átomos primitivos (esquerda). Então, selecionando dois átomos âncora das duas estruturas primitivas a serem comparadas (no meio, em verde e turquesa), um número entre 0 e 1 pode ser calculado usando a fórmula vista acima. Crédito:Universidade Eötvös Loránd
Em um artigo publicado recentemente na Nature Communications , o Grupo de Pesquisa de Modelagem de Proteínas HUN-REN-ELTE (Instituto de Química) lançou as bases para um método matemático, permitindo a comparação assistida por computador das estruturas tridimensionais das proteínas. O método é único porque, embora as alternativas disponíveis até agora levassem em consideração apenas a posição dos átomos, a nova técnica, chamada LoCoHD (Local Composition Hellinger Distance), também inclui as informações químicas dos átomos.