Pesquisadores aplicam métodos de computação quântica à previsão da estrutura de proteínas
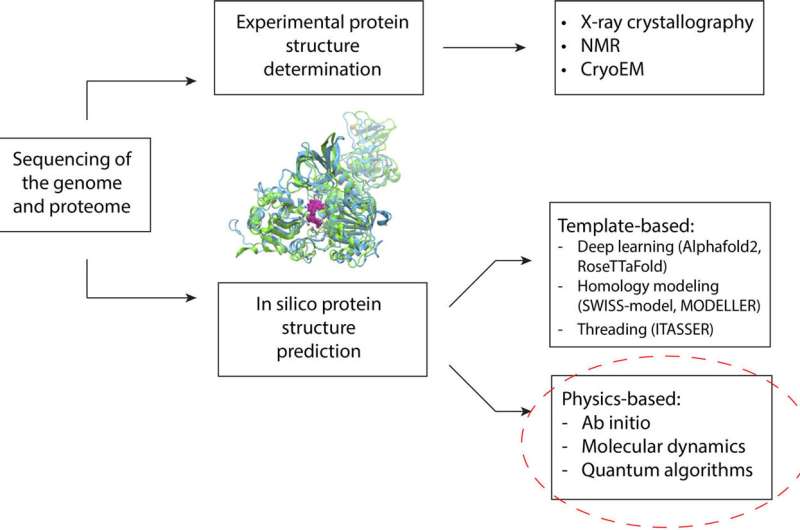 Visão geral do pipeline do PSP. Após o sequenciamento genômico, a sequência primária de aminoácidos é determinada. O método experimental começa então com a expressão desta proteína através da modificação genética de outro organismo com esta nova sequência. Este organismo irá então traduzir essas proteínas, e a nova proteína de interesse pode ser isolada, purificada e então resolvida usando cristalografia de raios X, RMN ou CryoEM. Os métodos in silico, por outro lado, simplesmente tomam a sequência primária de aminoácidos como entrada e a estrutura é prevista por um método baseado na física (onde a biofísica subjacente é de alguma forma simulada) ou um método baseado em modelo (onde algoritmos de aprendizado de máquina prever estruturas com base em padrões encontrados em um conjunto de treinamento de modelos experimentais). O método que adotamos neste trabalho se enquadra na categoria de algoritmos baseados em física. Como exemplo ilustrado, um modelo in silico e uma estrutura cristalina de raios X da helicase SARS-CoV2 NSP13 (PDB:7NN0) são sobrepostos, juntamente com um inibidor conhecido acoplado (colorido em magenta). Crédito:Journal of Chemical Theory and Computation (2024). DOI:10.1021/acs.jctc.4c00067
Visão geral do pipeline do PSP. Após o sequenciamento genômico, a sequência primária de aminoácidos é determinada. O método experimental começa então com a expressão desta proteína através da modificação genética de outro organismo com esta nova sequência. Este organismo irá então traduzir essas proteínas, e a nova proteína de interesse pode ser isolada, purificada e então resolvida usando cristalografia de raios X, RMN ou CryoEM. Os métodos in silico, por outro lado, simplesmente tomam a sequência primária de aminoácidos como entrada e a estrutura é prevista por um método baseado na física (onde a biofísica subjacente é de alguma forma simulada) ou um método baseado em modelo (onde algoritmos de aprendizado de máquina prever estruturas com base em padrões encontrados em um conjunto de treinamento de modelos experimentais). O método que adotamos neste trabalho se enquadra na categoria de algoritmos baseados em física. Como exemplo ilustrado, um modelo in silico e uma estrutura cristalina de raios X da helicase SARS-CoV2 NSP13 (PDB:7NN0) são sobrepostos, juntamente com um inibidor conhecido acoplado (colorido em magenta). Crédito:Journal of Chemical Theory and Computation (2024). DOI:10.1021/acs.jctc.4c00067
Pesquisadores da Cleveland Clinic e da IBM publicaram recentemente descobertas no Journal of Chemical Theory and Computation isso poderia estabelecer as bases para a aplicação de métodos de computação quântica à previsão da estrutura de proteínas.