Metas de IDs da estrutura de aprendizado de máquina para melhorar os catalisadores
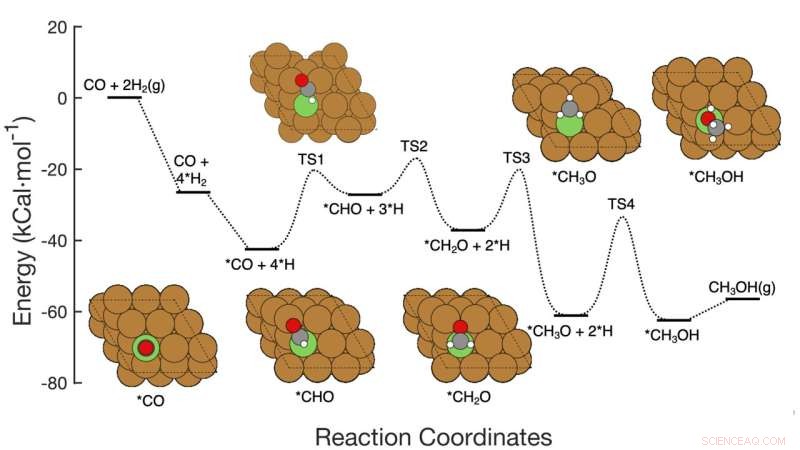
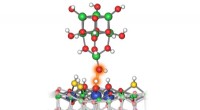
Este gráfico mostra a via de reação de sete etapas da hidrogenação de CO para metanol sobre catalisadores à base de cobre, incluindo os reagentes em cada etapa, arranjos atômicos esquemáticos dos intermediários e as barreiras de ativação de energia necessárias para passar de uma etapa a outra. A equipe do Brookhaven Lab demonstrou uma estrutura de aprendizado de máquina que identificou com sucesso quais etapas/combinações de etapas devem ser ajustadas para melhorar a produção de metanol. Seu trabalho pode ajudar a orientar o projeto de novos catalisadores para atingir esse objetivo e a estrutura pode ser aplicada para otimizar outras reações. Crédito:Laboratório Nacional de Brookhaven
Químicos do Laboratório Nacional Brookhaven do Departamento de Energia dos EUA desenvolveram uma nova estrutura de aprendizado de máquina (ML) que pode se concentrar em quais etapas de uma conversão química de várias etapas devem ser ajustadas para melhorar a produtividade. A abordagem pode ajudar a orientar o projeto de catalisadores - "negociantes" químicos que aceleram as reações.
A equipe desenvolveu o método para analisar a conversão de monóxido de carbono (CO) em metanol usando um catalisador à base de cobre. A reação consiste em sete passos elementares bastante simples.
"Nosso objetivo era identificar qual etapa elementar na rede de reação ou qual subconjunto de etapas controla a atividade catalítica", disse Wenjie Liao, o primeiro autor de um artigo que descreve o método recém publicado na revista
Catalysis Science &Technology . Liao é um estudante de pós-graduação na Stony Brook University que vem trabalhando com cientistas do grupo de Reatividade e Estrutura de Catálise (CRS) na Divisão de Química do Brookhaven Lab.
Ping Liu, o químico do CRS que liderou o trabalho, disse:"Usamos essa reação como um exemplo de nosso método de estrutura de ML, mas você pode colocar qualquer reação nessa estrutura em geral".
Direcionar energias de ativação Imagine uma reação química de várias etapas como uma montanha-russa com colinas de diferentes alturas. A altura de cada colina representa a energia necessária para ir de um degrau ao outro. Os catalisadores reduzem essas "barreiras de ativação", facilitando a união dos reagentes ou permitindo que eles o façam em temperaturas ou pressões mais baixas. Para acelerar a reação geral, um catalisador deve direcionar a etapa ou etapas que têm o maior impacto.
Tradicionalmente, os cientistas que buscam melhorar essa reação calculam como alterar cada barreira de ativação
uma de cada vez pode afetar a taxa de produção global. Esse tipo de análise pode identificar qual etapa foi "limitante de velocidade" e quais etapas determinam a seletividade da reação - isto é, se os reagentes prosseguem para o produto desejado ou seguem um caminho alternativo para um subproduto indesejado.

O químico do Brookhaven Lab, Ping Liu, e Wenjie Liao, estudante de pós-graduação da Stony Brook University, desenvolveram uma estrutura de aprendizado de máquina para identificar quais etapas de reação química podem ser direcionadas para melhorar a produtividade da reação. Crédito:Laboratório Nacional de Brookhaven
Mas, de acordo com Liu, "essas estimativas acabam sendo muito grosseiras com muitos erros para alguns grupos de catalisadores. Isso prejudicou muito o projeto e a triagem de catalisadores, que é o que estamos tentando fazer", disse ela.
A nova estrutura de aprendizado de máquina foi projetada para melhorar essas estimativas para que os cientistas possam prever melhor como os catalisadores afetarão os mecanismos de reação e a produção química.
"Agora, em vez de mover uma barreira de cada vez, estamos movendo todas as barreiras simultaneamente. E usamos aprendizado de máquina para interpretar esse conjunto de dados", disse Liao.
Essa abordagem, disse a equipe, fornece resultados muito mais confiáveis, inclusive sobre como as etapas de uma reação funcionam juntas.
"Sob condições de reação, essas etapas não são isoladas ou separadas umas das outras; estão todas conectadas", disse Liu. "Se você fizer um passo de cada vez, perderá muitas informações - as interações entre as etapas elementares. Isso é o que foi capturado neste desenvolvimento", disse ela.
Construindo o modelo Os cientistas começaram construindo um conjunto de dados para treinar seu modelo de aprendizado de máquina. O conjunto de dados foi baseado em cálculos da "teoria do funcional da densidade" (DFT) da energia de ativação necessária para transformar um arranjo de átomos no próximo através das sete etapas da reação. Em seguida, os cientistas executaram simulações em computador para explorar o que aconteceria se eles mudassem todas as sete barreiras de ativação simultaneamente – algumas subindo, outras descendo, algumas individualmente e algumas em pares.
"A gama de dados que incluímos foi baseada na experiência anterior com essas reações e esse sistema catalítico, dentro da interessante faixa de variação que provavelmente proporcionará melhor desempenho", disse Liu.
Ao simular variações em 28 "descritores" - incluindo as energias de ativação para as sete etapas mais pares de etapas mudando duas de cada vez - a equipe produziu um conjunto de dados abrangente de 500 pontos de dados. Esse conjunto de dados previu como todos esses ajustes individuais e pares de ajustes afetariam a produção de metanol. O modelo então classificou os 28 descritores de acordo com sua importância na condução da produção de metanol.
"Nosso modelo 'aprendeu' com os dados e identificou seis descritores-chave que ele prevê que teriam o maior impacto na produção", disse Liao.
Depois que os descritores importantes foram identificados, os cientistas treinaram novamente o modelo de ML usando apenas esses seis descritores "ativos". Esse modelo de ML aprimorado foi capaz de prever a atividade catalítica com base puramente em cálculos de DFT para esses seis parâmetros.
"Ao invés de você ter que calcular todos os 28 descritores, agora você pode calcular com apenas os seis descritores e obter as taxas de conversão de metanol que você está interessado", disse Liu.
A equipe diz que também pode usar o modelo para selecionar catalisadores. Se eles puderem projetar um catalisador que melhore o valor dos seis descritores ativos, o modelo prevê uma taxa máxima de produção de metanol.
Compreendendo os mecanismos Quando a equipe comparou as previsões de seu modelo com o desempenho experimental de seu catalisador – e o desempenho de ligas de vários metais com cobre – as previsões coincidiram com as descobertas experimentais. As comparações da abordagem ML com o método anterior usado para prever o desempenho das ligas mostraram que o método ML é muito superior.
Os dados também revelaram muitos detalhes sobre como as mudanças nas barreiras de energia podem afetar o mecanismo de reação. De particular interesse - e importância - foi como as diferentes etapas da reação funcionam juntas. Por exemplo, os dados mostraram que, em alguns casos, a redução da barreira de energia na etapa de limitação de taxa por si só não melhoraria a produção de metanol. Mas ajustar a barreira de energia de uma etapa anterior na rede de reação, mantendo a energia de ativação da etapa limitante de taxa dentro de uma faixa ideal, aumentaria a produção de metanol.
"Nosso método nos fornece informações detalhadas que podemos usar para projetar um catalisador que coordene bem a interação entre essas duas etapas", disse Liu.
Mas Liu está mais empolgado com o potencial de aplicar essas estruturas de ML orientadas por dados a reações mais complicadas.
"Usamos a reação do metanol para demonstrar nosso método. Mas a maneira como ele gera o banco de dados e como treinamos o modelo de ML e como interpolamos o papel de cada função do descritor para determinar o peso geral em termos de importância - isso pode ser aplicado a outras reações facilmente", disse ela.
+ Explorar mais Descoberta de um novo catalisador para hidrogenação de dióxido de carbono altamente ativa e seletiva em metanol