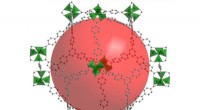
Específico de sequência, induzida por torção, configurações elásticas dobradas, gerado por simulações de dinâmica molecular em supercomputadores no Texas Advanced Computing Center, ajudam a explicar como longas fitas de DNA podem caber em espaços pequenos. Crédito:Christopher G. Myers, B. Montgomery Pettitt, University of Texas Medical Branch
Um mistério biológico está no centro de cada uma de nossas células, a saber:como um metro de DNA pode ser empacotado no espaço de um mícron (ou um milionésimo de metro) dentro de cada núcleo do nosso corpo.
Os núcleos das células humanas não são nem mesmo o lugar biológico mais populoso que conhecemos. Alguns bactiófagos - vírus que infectam e se replicam dentro de uma bactéria - têm DNA ainda mais concentrado.
"Como é que entra aí?" B. Montgomery (Monte) Pettitt, um bioquímico e professor da University of Texas Medical Branch, pergunta. "É um polímero carregado. Como ele supera a repulsão em sua densidade líquido-cristalina? Quanta ordem e desordem são permitidas, e como isso desempenha um papel nos ácidos nucléicos? "
Usando os supercomputadores Stampede e Lonestar5 da Universidade do Texas no Texas Advanced Computing Center (TACC) de Austin, Pettitt investiga como o DNA dos fagos se dobra em espaços hiper-confinados.
Escrito na edição de junho de 2017 da Journal of Computational Chemistry , ele explicou como o DNA pode superar a repulsão eletrostática e sua rigidez natural.
A chave para fazer isso? Kinks.
A introdução de torções ou curvas acentuadas em configurações de DNA empacotado dentro de um envelope esférico reduz significativamente as energias e pressões gerais da molécula, de acordo com Pettitt.
Ele e seus colaboradores usaram um modelo que deforma e torce o DNA a cada 24 pares de bases, que está próximo do comprimento médio que é previsto a partir da sequência de DNA do fago. A introdução de tais defeitos persistentes não apenas reduz a energia de dobra total do DNA confinado, mas também reduz o componente eletrostático da energia e pressão.
"Mostramos que um amplo conjunto de configurações de polímero é consistente com os dados estruturais, ele e o colaborador Christopher Myers, também da University of Texas Medical Branch, escreveu.
Insights como esses não podem ser obtidos estritamente no laboratório. Eles exigem supercomputadores que funcionam como microscópios moleculares, mapear o movimento dos átomos e ligações atômicas em escalas de comprimento e tempo que não são viáveis para estudar apenas com experimentos físicos.
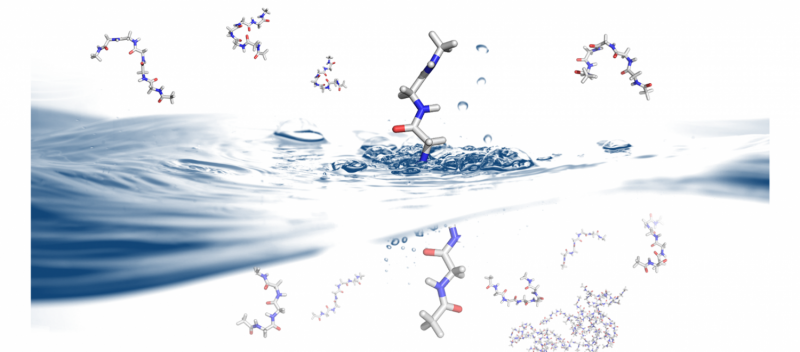
Como e por que as proteínas se dobram é um problema que tem implicações para o projeto e a terapêutica das proteínas. B. Montgomery Pettitt e seu grupo de pesquisa na University of Texas Medical Branch usam os supercomputadores Stampede e Lonestar5 no Texas Advanced Computing Center para explorar a dinâmica do dobramento de proteínas em solução. Crédito:Christopher G. Myers, B. Montgomery Pettitt, University of Texas Medical Branch
“No campo da biologia molecular, há uma interação maravilhosa entre a teoria, experimento e simulação, "Pettitt disse." Nós pegamos parâmetros de experimentos e vemos se eles concordam com as simulações e teorias. Este se torna o método científico de como agora avançamos nossas hipóteses. "
Problemas como aqueles em que Pettitt está interessado não podem ser resolvidos em um computador desktop ou em um cluster típico de campus, mas requerem centenas de processadores de computador trabalhando em paralelo para imitar os movimentos mínimos e as forças físicas das moléculas em uma célula.
Pettitt é capaz de acessar os supercomputadores do TACC em parte por causa de um programa único conhecido como Journal of Computational Chemistry iniciativa, o que torna os recursos de computação do TACC, expertise e treinamento disponíveis para pesquisadores nas 14 instituições da University of Texas Systems.
"Pesquisa computacional, como o do Dr. Pettitt, que busca unir nosso entendimento do físico, químico, e, finalmente, fenômenos biológicos, envolve tantos cálculos que só é realmente acessível em grandes supercomputadores como os sistemas TACC's Stampede ou Lonestar5, "disse Brian Beck, pesquisador de ciências da vida na TACC.
"Ter recursos de supercomputação TACC disponíveis é fundamental para este estilo de pesquisa, "Pettitt disse.
ENCONTRANDO A ORDEM EM PROTEÍNAS DESORDENADAS
Outro fenômeno que há muito tempo interessa a Pettitt é o comportamento de proteínas intrinsecamente desordenadas (IDPs) e domínios intrinsecamente desordenados, onde partes de uma proteína têm uma forma desordenada.
Ao contrário dos cristais ou do DNA altamente compactado nos vírus, que tem distintos, formas rígidas, IDPs "dobram-se em uma bagunça pegajosa, "de acordo com Pettitt. E ainda assim eles são essenciais para todas as formas de vida.
Acredita-se que em eucariotos (organismos cujas células têm subestruturas complexas como núcleos), cerca de 30 por cento das proteínas têm um domínio intrinsecamente desordenado. Mais de 60 por cento das proteínas envolvidas na sinalização celular (processos moleculares que recebem sinais de fora da célula ou através das células que dizem à célula quais comportamentos ativar e desativar em resposta) têm domínios desordenados. De forma similar, 80 por cento das proteínas de sinalização relacionadas ao câncer têm regiões IDP - tornando-as moléculas importantes para serem entendidas.
Entre os deslocados internos que Pettitt e seu grupo estão estudando estão os fatores de transcrição nuclear. Essas moléculas controlam a expressão de genes e têm um domínio de sinalização rico em aminoácidos flexíveis, glicina.

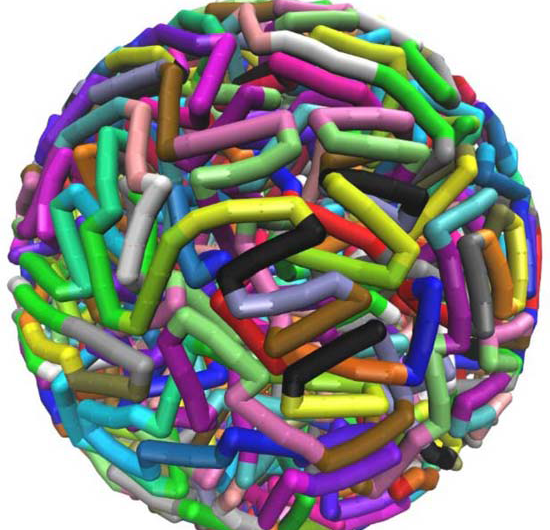
As imagens acima mostram as distribuições de densidade média em 21 configurações de DNA, cada uma simulada por 100 nanossegundos de dinâmica molecular após a minimização usando a) configurações totalmente elásticas eb) configurações dobradas, para comparação com c) mapa de densidade Cryo-EM de reconstruções de fago assimétricas de P22 com densidade de capsídeo removida graficamente. Crédito:Christopher G. Myers, B. Montgomery Pettitt, University of Texas Medical Branch
O dobramento do domínio de sinalização do fator de transcrição nuclear não é causado por ligações de hidrogênio e efeitos hidrofóbicos, como a maioria das moléculas de proteína, de acordo com Pettitt. Em vez, quando as moléculas mais longas encontram muitas glicinas em um espaço, eles vão além de sua solubilidade e começam a se associar de maneiras incomuns.
"É como adicionar muito açúcar ao seu chá, "Pettitt explica." Não vai ficar mais doce. O açúcar deve cair da solução e encontrar um parceiro - precipitando em um amontoado. "
Escrevendo em Ciência de Proteínas em 2015, ele descreveu simulações moleculares realizadas em Stampede que ajudaram a explicar como e por que IDPs colapsam em estruturas semelhantes a glóbulos.
As simulações calcularam as forças das interações dipolo-dipolo carbonil (CO) - atrações entre a extremidade positiva de uma molécula polar e a extremidade negativa de outra molécula polar. Ele determinou que essas interações são mais importantes no colapso e agregação de longas fitas de glicina do que na formação de ligações H.
"Dado que o backbone é uma característica de todas as proteínas, As interações de CO também podem desempenhar um papel em proteínas de sequência não trivial, onde a estrutura é eventualmente determinada por empacotamento interno e os efeitos estabilizadores de ligações H e interações CO-CO, "concluiu.
A pesquisa foi habilitada por uma alocação de tempo de computação no Stampede por meio do Extreme Science and Engineering Discovery Environment (XSEDE), que é apoiado pela National Science Foundation.
Pettitt, um campeão de supercomputação de longa data, não usa apenas os recursos do TACC. Ele encoraja outros estudiosos, incluindo seus colegas do Centro Sealy de Biologia Estrutural e Biofísica Molecular, para usar supercomputadores também.
"A computação avançada é importante para análise e refinamento de dados de experimentos, Raios-X e microscopia eletrônica, e informática, "ele diz." Todos esses problemas têm problemas de processamento de big data que podem ser resolvidos usando a computação avançada. "
Quando se trata de descobrir os mistérios da biologia nas menores escalas, nada supera um supercomputador gigante.