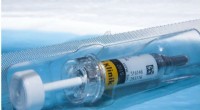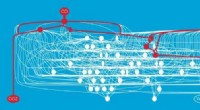
Animação mostrando como a proteína do pico do coronavírus muda sua forma pouco antes de se ligar ao receptor da célula humana. Crédito:Ilustração fornecida por Mahmoud Moradi.
O químico computacional Mahmoud Moradi desenvolverá aprimorado, Simulações 3-D da dinâmica molecular de glicoproteínas de pico de coronavírus para obter uma melhor compreensão de como o vírus se liga às células humanas.
O mapeamento de como essas proteínas sofrem mudanças conformacionais para se ligar aos receptores da célula hospedeira é crítico para o desenvolvimento de vacinas e terapêuticas de coronavírus. As simulações são especialmente importantes porque uma estrutura para o projeto de drogas exigirá dinâmica, visualizações tridimensionais de estruturas celulares e comportamento, em vez de uma imagem estática.
"Tal como acontece com outros vírus, uma etapa crucial no processo de infecção por coronavírus é a entrada viral, "disse Moradi, professor assistente na Faculdade de Artes e Ciências J. William Fulbright. "Com coronavírus, sabemos que essas glicoproteínas de pico medeiam a entrada na célula humana. Ambos SARS-CoV-2, a causa do COVID-19, e SARS-CoV, a causa da epidemia de SARS de 2002-2003, têm proteínas de pico que se ligam ao mesmo receptor nas células humanas. "
O trabalho de Moradi faz parte do COVID-19 High Performance Computing Consortium, uma colaboração do governo, parceiros da indústria e acadêmicos com foco em recursos de computação para pesquisas COVID-19. Liderado pelo Escritório de Política de Ciência e Tecnologia da Casa Branca, o Departamento de Energia dos EUA e IBM, o consórcio oferece tempo de computação e recursos gratuitos em alguns dos supercomputadores mais poderosos do mundo.
Para realizar as simulações, Moradi recebeu acesso a Frontera, um supercomputador patrocinado pela National Science Foundation, alojado na Universidade do Texas em Austin. Frontera é o maior supercomputador em qualquer campus universitário.
O projeto de Moradi se beneficia de vários modelos 3D de alta resolução das proteínas de pico do coronavírus. Esses modelos podem ser usados como estruturas iniciais para iniciar as simulações que permitirão a análise dos mecanismos detalhados das proteínas e seu comportamento na entrada viral. Melhorada, simulações detalhadas de tal dinâmica molecular fornecerão um quadro completo das mudanças estruturais das proteínas, bem como como eles se ligam à enzima conversora de angiotensina 2, o receptor específico da célula humana.
A pesquisa de Moradi encontra-se na interseção da biologia, física, química, matemática, estatística e ciência da computação. Suas simulações biomoleculares e teorias computacionais explicam como as proteínas, as moléculas burras de carga das células, função a nível molecular. Seu trabalho aprimora modelos geométricos para descrever como as proteínas mudam sua forma e como essas mudanças afetam o comportamento de uma proteína. Em fevereiro, ele recebeu $ 650, Prêmio 000 do Desenvolvimento no Início da Carreira do Corpo Docente da National Science Foundation por este trabalho.