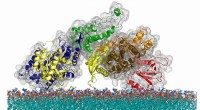
Os pesquisadores da Rice University foram inspirados pelo antigo trabalho dos ferreiros enquanto refinavam seus modelos computacionais de como as proteínas se dobram. Os modelos destinam-se a ajudar biólogos estruturais que projetam drogas e outras terapias. Crédito:Rice University / Wikipedia
Onde a ferraria da aldeia ficava agora existe um algoritmo, seu poderoso martelo matemático dando forma às proteínas.
A profissão de ferreiro é uma analogia válida para o que os cientistas da Rice University criaram:um novo método para fazer modelos estruturais precisos de proteínas que exige muito menos poder computacional do que as abordagens de força bruta existentes.
O objetivo dos modelos estruturais produzidos por computação, de acordo com o físico Peter Wolynes, do Centro de Física Teórica Biológica de Rice (CTBP), deve ser tão detalhado e útil quanto aqueles produzidos por laboriosos meios experimentais, particularmente cristalografia de raios-X, que fornecem localizações detalhadas para cada átomo dentro de uma proteína.
O novo método é inspirado na metalurgia. Como o ferreiro que deve não apenas aquecer e resfriar um metal, mas também bater no metal certo para movê-lo para mais perto de um produto útil, o projeto Rice, liderado por Wolynes e ex-aluno Xingcheng Lin, aplica força em pontos estratégicos durante a simulação de modelos de proteínas para acelerar o cálculo.
"Uma grande questão é se poderíamos nos tornar mais confiantes na precisão dos resultados de uma simulação do que nos resultados de experimentos de raios-X, "Wolynes disse." Estou prestes a dizer que é onde estamos agora, mas, claro, o tempo vai dizer."
O estudo aparece esta semana no Proceedings of the National Academy of Sciences .Os pesquisadores têm usado a cristalografia de raios-X por mais de um século para aprender as posições dos átomos dentro das moléculas a partir de suas estruturas nos cristais de proteínas. Essas informações são o ponto de partida para estudos de biologia estrutural, e a precisão é considerada essencial para projetar drogas para interagir com proteínas específicas.
Mas as estruturas cristalinas fornecem apenas um instantâneo de uma proteína que, na realidade, muda sua forma global e posições atômicas detalhadas à medida que a proteína realiza seu trabalho na célula.
Wolynes e seus colegas há muito são os pioneiros nos métodos computacionais para prever estruturas dobradas do cenário de energia codificado nos aminoácidos da proteína. No novo trabalho, eles tratam da colocação detalhada das cadeias laterais dos aminoácidos que podem ser empurrados desta ou daquela maneira por um algoritmo que começa a partir de uma visão de resolução moderada da estrutura global.
"Para alcançar a resolução, queremos começar com os modelos iniciais de granulação grossa, normalmente precisaríamos executar o computador por dois meses, "disse ele." Mas descobrimos que poderíamos primeiro simular os movimentos do modelo de granulação grossa para encontrar os movimentos que mudariam os padrões de ligação na molécula de forma mais substancial.
"Alguns movimentos não fazem nada:você pode estar batendo a mão, mas o movimento importante é dobrar o cotovelo, "Wolynes disse." Então, criamos uma receita para selecionar os movimentos mais significativos e os usamos para influenciar outra simulação feita em alta resolução. Nós usamos deliberadamente a força para empurrar as proteínas apenas nessas direções, em seguida, olhei para as estruturas resultantes para ver se eram mais estáveis do que quando começamos. "
Como um ferreiro martelando areia de um pedaço de metal, a equipe do Rice também encontrou métodos para eliminar a "areia" de seus modelos:movimento lento, cadeias laterais volumosas, cuja dinâmica lenta consumia o tempo do computador enquanto uma proteína se dobrava. Tirar a areia não mudou o resultado, mas tornou a computação muito mais rápida.
"Os metalúrgicos aquecem e resfriam as coisas para recozê-las, mas também descobrem como fazer grandes movimentos que não vão acontecer espontaneamente se você apenas mantiver o metal em alta temperatura, "Wolynes disse." Há muito tempo fazemos recozimento com modelos de granulação grossa. Mas os ferreiros também martelam o metal para tirar a areia, ou escória, e isso nos inspirou a deformar proteínas mecanicamente, também."
Wolynes disse que o CTBP atualizou metodicamente seus modelos de dobramento de proteínas e previsão de estrutura usando novas linguagens de computador ao longo dos anos, o que, por sua vez, ajudou os pesquisadores a atacar problemas mais sofisticados.
"A recodificação dos modelos nos permitiu olhar para moléculas que são 10 vezes maiores do que antes, "disse ele." Não há nova física, apenas nova programação e melhores computadores paralelos, mas está fazendo uma diferença real nos problemas práticos que agora podemos enfrentar. "