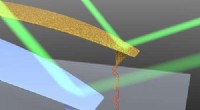
A maioria dos métodos para a caracterização estrutural de biomoléculas, como cristalografia de raios-X ou microscopia eletrônica, requerem amostras estáticas ou cristalizadas. Anexar moléculas fluorescentes a superfícies de proteínas, Contudo, permite a imagem direta de interações biomoleculares dinâmicas usando luz, que poderia ser melhorado, digamos pesquisadores A * STAR, com modelagem preditiva de tempos de vida de fluorescência.
A fluorescência normalmente envolve moléculas únicas que absorvem luz espontaneamente e depois a reemitem com uma cor diferente. Mas nas condições certas, um fóton absorvido pode pular de uma molécula doadora para um composto aceitador próximo que também sofre fluorescência. Pesquisadores exploraram recentemente a forte dependência de distância desse efeito para produzir 'réguas espectroscópicas' que medem a dinâmica em nanoescala entre sondas doadoras e aceitadoras anexadas a diferentes partes de uma estrutura protéica.
Um dos principais desafios é fazer réguas espectroscópicas com precisão aceitável. Os fluoróforos convencionais têm grandes, estruturas flexíveis que pressionam contra as proteínas de várias maneiras, tornando difícil medir o comprimento da régua. Então, para buscar alternativas, Tsz Sian Chwee e colegas de trabalho do Instituto A * STAR de Computação de Alto Desempenho investigaram se podiam calcular a fluorescência de moléculas rígidas e pequenas conhecidas como sin-bimanes, e então usar essas teorias para o design da sonda.
Abordagens típicas da química quântica, Contudo, têm problemas para calcular as propriedades quando uma molécula absorve um fóton e entra em um estado excitado. Chwee e sua equipe esperavam superar essas imprecisões usando a teoria do funcional da densidade dependente do tempo, que trata o problema dos elétrons excitados com um algoritmo de 'correlação de troca' derivado parcialmente de experimentos.
"A teoria do funcional da densidade dependente do tempo é usada pela comunidade científica para estudar fenômenos como absorção e emissão, mas todo o potencial desta abordagem ainda não foi aproveitado, "diz Chwee.
Usando tempos de vida de fluorescência como um parâmetro de teste, os pesquisadores compararam como diferentes teorias de correlação de troca simularam sin-bimanes em realismo, situações cheias de solvente. Esses testes revelaram que os modelos que incorporam interações vibrônicas - o acoplamento sincronizado de vibrações moleculares a excitações eletrônicas - fornecem as previsões mais precisas de tempos de vida fluorescentes. Eles descobriram várias funções de correlação de troca que são capazes de lidar com essas equações com um custo computacional mínimo.
"Aspectos vibrônicos foram amplamente esquecidos, embora desempenhem papéis decisivos na fotofísica de moléculas fluorescentes, "observa Chwee." Enquanto realizávamos nossos cálculos em supercomputadores, os recursos computacionais são modestos o suficiente para serem concluídos em uma estação de trabalho moderna em algumas semanas. "
Chwee antecipa que a análise rápida usando teorias do funcional da densidade pode ser melhor para detectar candidatos raros a sondas fluorescentes com forte absorção e propriedades de emissão ajustáveis.