Os bioinformáticos eliminam uma etapa desnecessária na análise de estabilidade de proteínas
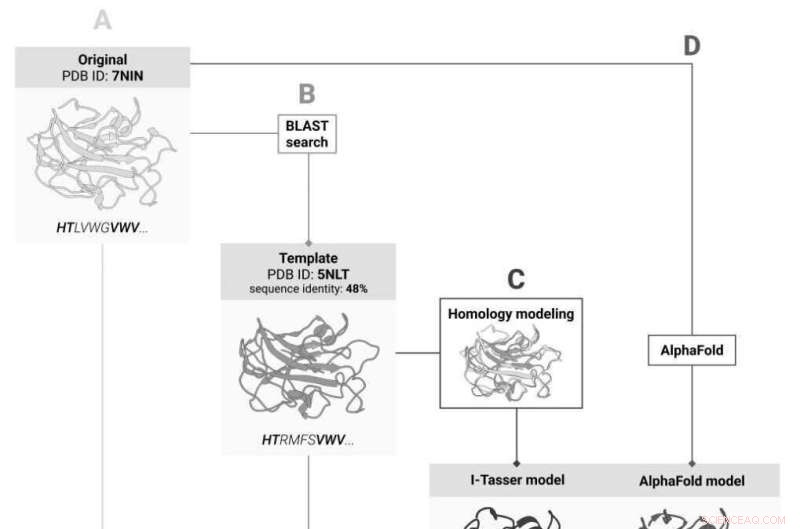
Quatro maneiras de prever mudanças na estabilidade da proteína após a mutação:(A) pela estrutura da proteína original; (B) pela estrutura do seu homólogo; (C) pela estrutura da proteína original prevista com base na estrutura do holomlog, e (D) pela estrutura prevista pela inteligência artificial com base na sequência de aminoácidos. Crédito:Instituto de Ciência e Tecnologia Skolkovo
Pesquisadores do Centro Skoltech de Biologia Molecular e Celular compararam diferentes métodos de previsão de estrutura de proteínas em termos de avaliação da estabilidade de proteínas mutantes e obtiveram o mesmo resultado para as estruturas previstas por IA e tridimensionais experimentais (3D) de proteínas com sequências de aminoácidos semelhantes. No entanto, a tentativa de prever a estrutura da proteína alvo a partir da estrutura conhecida de seu "parente" apenas tornou a previsão menos precisa. As descobertas da equipe facilitarão os cálculos preliminares na avaliação das alterações de estabilidade causadas pela mutação. A pesquisa foi publicada em
Bioinformática .
Experimentos biológicos geralmente envolvem proteínas mutantes, que são necessárias para o estudo da estrutura e funções da proteína ou processos celulares, bem como engenharia de proteínas. Mutações são conhecidas por afetar a estrutura e estabilidade de uma proteína. Como os experimentos são muito caros e demorados, os cientistas estão criando uma solução alternativa na forma de métodos computacionais para avaliar o impacto das mutações na estabilidade. No entanto, suas aplicações exigem o conhecimento da estrutura 3D de uma proteína.
Uma estrutura 3D experimental não está disponível para todas as proteínas e é provável que esteja faltando para aquela visada pela equipe. Se este for o caso, os modelos 3D dos homólogos da proteína, ou seja, seus "parentes mais próximos", podem fornecer a linha de vida, porque o grau de semelhança nas sequências de aminoácidos que garante uma boa correspondência entre as estruturas 3D das proteínas é bem conhecido. A solução seria primeiro prever a estrutura da proteína com base na estrutura conhecida de seu homólogo e então calcular o impacto das mutações para o modelo previsto.
Graças ao avanço do ano passado na previsão da estrutura da proteína, os cientistas agora têm uma alternativa:em vez de prever a estrutura 3D com base em homólogos, eles podem usar a ferramenta AlphaFold baseada em IA, que prevê a estrutura da proteína a partir da sequência de aminoácidos e já tratou com a grande maioria das proteínas conhecidas até o momento.
Em seu estudo recente, os pesquisadores da Skoltech decidiram descobrir qual dessas abordagens funciona melhor para prever mudanças de estabilidade após mutação. Por mais preciso que o AlphaFold possa ser, encontrar a estrutura da proteína por meio de experimentos ainda continua sendo o "padrão ouro". Ao comparar as duas abordagens, a equipe usou sete métodos de avaliação de estabilidade e comparou seus resultados com os de AlphaFold e I-Tasser, o melhor sistema de previsão de estrutura baseado em homólogos. Além disso, os pesquisadores verificaram se podem pular a previsão de estrutura baseada em homólogos e calcular a estabilidade para a estrutura conhecida da proteína homóloga.
"Decidimos descobrir até que ponto divergiríamos da previsão precisa se usássemos a estrutura da proteína 'vizinha' em vez da real. Descobrimos que a etapa de previsão baseada em homologia só piora as coisas ao produzir um resultado menos preciso. Mostramos que praticamente não faz diferença se você usa a estrutura experimental do homólogo ou a previsão do AlphaFold. De certa forma, tratava-se de validação:quando confrontado com um novo método, você deve verificar se ele funciona para sua tarefa em primeiro lugar . Isso é exatamente o que fizemos", primeiro autor do estudo, Skoltech Ph.D. aluna Marina Pak, comenta.
"Com todo esse alarido sobre o AlphaFold, alguns cientistas e não profissionais acreditam que ele resolveu todos os problemas de pesquisa de proteínas em biologia computacional, mas não. Por exemplo, a previsão de mudanças de estabilidade induzidas por mutações ainda apresenta baixa confiabilidade, mesmo embora a mudança na estabilidade esteja entre os principais impulsionadores da funcionalidade da proteína. Uma ferramenta que pudesse determinar inequivocamente o impacto da mutação na estabilidade ajudaria tanto no planejamento do experimento quanto na interpretação dos resultados. Suponha que, para uma proteína que não seja ótima em termos de estabilidade, desejamos encontrar mutações que a tornem estável nas condições desejadas, por exemplo, para garantir que ela permaneça ativa em alta temperatura. Uma vez que possamos fazer isso apenas por meio de cálculos, a abordagem de redesenho e otimização de proteínas mudará drasticamente " principal autor do estudo, conclui o professor assistente da Skoltech, Dmitry Ivankov.
Embora a previsão de mudança de estabilidade pareça mais fácil do que prever a estrutura 3D, ainda continua sendo um desafio intratável mesmo para a IA. Dados de treinamento escassos são apenas um dos problemas:o AlphaFold tinha quase 200.000 estruturas de proteínas para treinar, mas dados experimentais sobre mudanças de estabilidade somam milhares de conjuntos, cobrindo apenas algumas dezenas de proteínas únicas. Os autores esperam que, se mais dados estiverem disponíveis e os pesquisadores mostrarem maior interesse na tarefa, um avanço certamente acontecerá em breve.
+ Explorar mais Os físicos usam IA para encontrar os nós de proteína mais complexos até agora