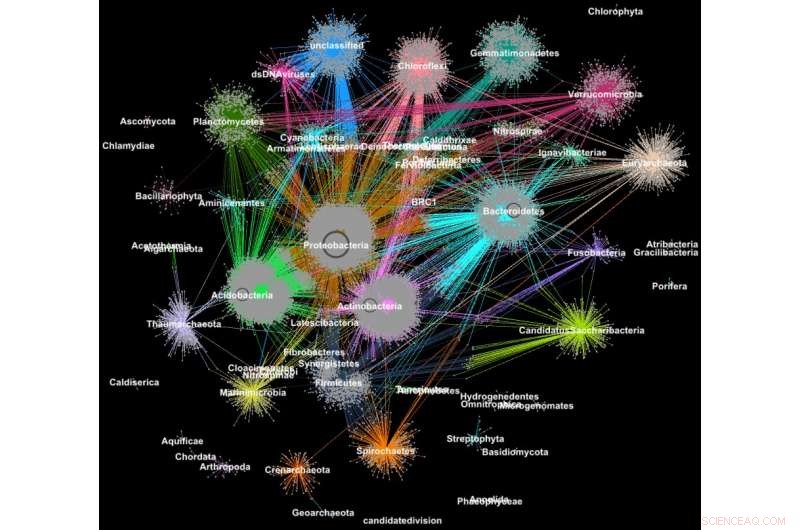
Proteínas de metagenomas agrupadas em famílias de acordo com sua classificação taxonômica. Crédito:Georgios Pavlopoulos e Nikos Kyrpides, JGI / Berkeley Lab
Você sabia que as ferramentas usadas para analisar as relações entre os usuários de redes sociais ou classificar as páginas da web também podem ser extremamente valiosas para compreender os dados de big science? Em uma rede social como o Facebook, cada usuário (pessoa ou organização) é representado como um nó e as conexões (relacionamentos e interações) entre eles são chamadas de arestas. Ao analisar essas conexões, os pesquisadores podem aprender muito sobre cada usuário - interesses, hobbies, hábitos de compra, amigos, etc.
Em biologia, algoritmos de agrupamento de grafos semelhantes podem ser usados para entender as proteínas que executam a maioria das funções vitais. Estima-se que o corpo humano sozinho contém cerca de 100, 000 tipos diferentes de proteínas, e quase todas as tarefas biológicas - da digestão à imunidade - ocorrem quando esses microrganismos interagem entre si. Uma melhor compreensão dessas redes pode ajudar os pesquisadores a determinar a eficácia de um medicamento ou identificar tratamentos potenciais para uma variedade de doenças.
Hoje, tecnologias avançadas de alto rendimento permitem que os pesquisadores capturem centenas de milhões de proteínas, genes e outros componentes celulares ao mesmo tempo e em uma variedade de condições ambientais. Algoritmos de agrupamento são então aplicados a esses conjuntos de dados para identificar padrões e relacionamentos que podem apontar para semelhanças estruturais e funcionais. Embora essas técnicas tenham sido amplamente utilizadas por mais de uma década, eles não conseguem acompanhar a torrente de dados biológicos gerados por sequenciadores e microarranjos de próxima geração. Na verdade, muito poucos algoritmos existentes podem agrupar uma rede biológica contendo milhões de nós (proteínas) e bordas (conexões).
É por isso que uma equipe de pesquisadores do Lawrence Berkeley National Laboratory (Berkeley Lab) e do Joint Genome Institute (JGI) do Departamento de Energia (DOE) adotou uma das abordagens de agrupamento mais populares da biologia moderna - o algoritmo Markov Clustering (MCL) - e modificou-o para ser executado rapidamente, com eficiência e em escala em supercomputadores de memória distribuída. Em um caso de teste, seu algoritmo de alto desempenho, chamado HipMCL, alcançou uma façanha anteriormente impossível:agrupar uma grande rede biológica contendo cerca de 70 milhões de nós e 68 bilhões de bordas em algumas horas, usando aproximadamente 140, 000 núcleos de processador no supercomputador Cori do National Energy Research Scientific Computing Center (NERSC). Um artigo que descreve este trabalho foi publicado recentemente na revista. Pesquisa de ácidos nucléicos .
"O benefício real do HipMCL é sua capacidade de agrupar redes biológicas massivas que eram impossíveis de agrupar com o software MCL existente, permitindo-nos identificar e caracterizar o novo espaço funcional presente nas comunidades microbianas, "diz Nikos Kyrpides, que chefia os esforços de Microbiome Data Science da JGI e o Prokaryote Super Program e é co-autor do artigo. "Além disso, podemos fazer isso sem sacrificar a sensibilidade ou precisão do método original, que é sempre o maior desafio nesse tipo de esforço de dimensionamento. "
"À medida que nossos dados crescem, está se tornando ainda mais imperativo que movamos nossas ferramentas para ambientes de computação de alto desempenho, "ele acrescenta." Se você me perguntasse qual é o tamanho do espaço das proteínas? A verdade é, não sabemos realmente porque até agora não tínhamos as ferramentas computacionais para efetivamente agrupar todos os nossos dados genômicos e sondar a matéria escura funcional. "
Além dos avanços na tecnologia de coleta de dados, os pesquisadores estão optando cada vez mais por compartilhar seus dados em bancos de dados comunitários, como o sistema Integrated Microbial Genomes &Microbiomes (IMG / M), que foi desenvolvido por meio de uma colaboração de décadas entre cientistas do JGI e da Divisão de Pesquisa Computacional (CRD) do Berkeley Lab. Mas, ao permitir que os usuários façam análises comparativas e explorem as capacidades funcionais das comunidades microbianas com base em sua sequência metagenômica, ferramentas da comunidade como IMG / M também estão contribuindo para a explosão de dados em tecnologia.
Como caminhadas aleatórias levam a gargalos de computação
Para controlar essa torrente de dados, pesquisadores contam com análise de cluster, ou agrupamento. Esta é essencialmente a tarefa de agrupar objetos de forma que os itens no mesmo grupo (cluster) sejam mais semelhantes do que aqueles em outros clusters. Por mais de uma década, biólogos computacionais têm favorecido o MCL para agrupar proteínas por semelhanças e interações.
"Uma das razões pelas quais o MCL se tornou popular entre os biólogos computacionais é que ele é relativamente livre de parâmetros; os usuários não precisam definir uma tonelada de parâmetros para obter resultados precisos e é notavelmente estável a pequenas alterações nos dados. importante porque você pode ter que redefinir uma semelhança entre os pontos de dados ou pode ter que corrigir um pequeno erro de medição em seus dados. Nesses casos, você não quer que suas modificações mudem a análise de 10 clusters para 1, 000 clusters, "diz Aydin Buluç, um cientista do CRD e um dos co-autores do artigo.
Mas, ele adiciona, a comunidade de biologia computacional está encontrando um gargalo de computação porque a ferramenta é executada principalmente em um único nó de computador, é computacionalmente caro para executar e tem uma grande pegada de memória - tudo isso limita a quantidade de dados que esse algoritmo pode agrupar.
Uma das etapas mais computacionalmente e de memória intensiva nesta análise é um processo chamado passeio aleatório. Esta técnica quantifica a força de uma conexão entre os nós, que é útil para classificar e prever links em uma rede. No caso de uma pesquisa na Internet, isso pode ajudá-lo a encontrar um quarto de hotel barato em San Francisco para as férias de primavera e até mesmo dizer a melhor época para reservá-lo. Em biologia, essa ferramenta pode ajudá-lo a identificar proteínas que estão ajudando seu corpo a combater o vírus da gripe.
Dado um gráfico ou rede arbitrária, é difícil saber a maneira mais eficiente de visitar todos os nós e links. Um passeio aleatório dá uma ideia da pegada, explorando todo o gráfico aleatoriamente; ele começa em um nó e se move arbitrariamente ao longo de uma aresta até um nó vizinho. Este processo continua até que todos os nós da rede de grafos tenham sido alcançados. Como existem muitas maneiras diferentes de viajar entre os nós de uma rede, esta etapa se repete inúmeras vezes. Algoritmos como o MCL continuarão executando esse processo de passeio aleatório até que não haja mais uma diferença significativa entre as iterações.
Em qualquer rede, você pode ter um nó conectado a centenas de nós e outro nó com apenas uma conexão. As caminhadas aleatórias irão capturar os nós altamente conectados porque um caminho diferente será detectado cada vez que o processo for executado. Com esta informação, o algoritmo pode prever com um nível de certeza como um nó da rede está conectado a outro. Entre cada corrida de passeio aleatório, o algoritmo marca sua previsão para cada nó do gráfico em uma coluna de uma matriz de Markov - uma espécie de livro-razão - e os clusters finais são revelados no final. Parece bastante simples, mas para redes de proteínas com milhões de nós e bilhões de arestas, isso pode se tornar um problema extremamente computacional e intensivo de memória. Com HipMCL, Os cientistas da computação do Berkeley Lab usaram ferramentas matemáticas de ponta para superar essas limitações.
"Temos notavelmente mantido o backbone MCL intacto, fazendo do HipMCL uma implementação massivamente paralela do algoritmo MCL original, "diz Ariful Azad, um cientista da computação no CRD e autor principal do artigo.
Embora tenha havido tentativas anteriores de paralelizar o algoritmo MCL para ser executado em uma única GPU, a ferramenta ainda só conseguia agrupar redes relativamente pequenas por causa das limitações de memória em uma GPU, Notas de Azad.
"Com o HipMCL, essencialmente retrabalhamos os algoritmos MCL para serem executados com eficiência, em paralelo em milhares de processadores, e configurá-lo para aproveitar a memória agregada disponível em todos os nós de computação, "ele acrescenta." A escalabilidade sem precedentes do HipMCL vem de seu uso de algoritmos de última geração para manipulação de matriz esparsa. "
Segundo Buluç, realizar um passeio aleatório simultaneamente a partir de muitos nós do gráfico é melhor calculado usando a multiplicação de matriz de matriz esparsa, que é uma das operações mais básicas no padrão GraphBLAS lançado recentemente. Buluç e Azad desenvolveram alguns dos algoritmos paralelos mais escaláveis para a multiplicação de matrizes esparsas do GraphBLAS e modificaram um de seus algoritmos de última geração para HipMCL.
"O ponto crucial aqui era encontrar o equilíbrio certo entre o paralelismo e o consumo de memória. O HipMCL extrai dinamicamente o máximo de paralelismo possível, dada a memória disponível alocada para ele, "diz Buluç.
HipMCL:Clustering em escala
Além das inovações matemáticas, outra vantagem do HipMCL é sua capacidade de funcionar perfeitamente em qualquer sistema, incluindo laptops, estações de trabalho e grandes supercomputadores. Os pesquisadores conseguiram isso desenvolvendo suas ferramentas em C ++ e usando bibliotecas MPI e OpenMP padrão.
"Testamos extensivamente o HipMCL no Intel Haswell, Processadores Ivy Bridge e Knights Landing na NERSC, usando um até 2, 000 nós e meio milhão de threads em todos os processadores, e em todas essas execuções, o HipMCL agrupou redes com sucesso compreendendo milhares a bilhões de arestas, "diz Buluç." Vemos que não há barreira no número de processadores que pode usar para ser executado e descobrimos que pode agrupar redes 1, 000 vezes mais rápido do que o algoritmo MCL original. "
"O HipMCL será realmente transformador para a biologia computacional de big data, assim como os sistemas IMG e IMG / M têm sido para a genômica do microbioma, "diz Kyrpides." Esta conquista é uma prova dos benefícios da colaboração interdisciplinar no Berkeley Lab. Como biólogos, entendemos a ciência, mas tem sido tão valioso poder colaborar com cientistas da computação que podem nos ajudar a enfrentar nossas limitações e nos impulsionar para frente. "
A próxima etapa é continuar a retrabalhar HipMCL e outras ferramentas de biologia computacional para futuros sistemas exascale, que será capaz de computar cálculos de quintilhões por segundo. Isso será essencial, pois os dados genômicos continuam a crescer a uma taxa alucinante - dobrando a cada cinco ou seis meses. Isso será feito como parte do centro de co-design Exagraph do DOE Exascale Computing Project.