O modelo de IA compara diretamente as propriedades de potenciais novos medicamentos
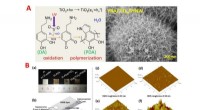
Arquiteturas tradicionais e de pares. A Os modelos tradicionais de aprendizado de máquina molecular recebem entradas moleculares singulares e prevêem propriedades absolutas das moléculas. As diferenças de propriedades previstas podem ser calculadas subtraindo os valores previstos para duas moléculas. Os modelos B Pairwise treinam nas diferenças nas propriedades de pares de moléculas para prever diretamente mudanças nas propriedades de derivatizações moleculares. As moléculas C são mescladas para criar pares somente após as divisões da validação cruzada para evitar o risco de vazamento de dados durante a avaliação do modelo. Portanto, cada molécula no conjunto de dados só pode ocorrer em pares nos dados de treinamento ou teste, mas não em ambos. Crédito:Journal of Cheminformatics (2023). DOI:10.1186/s13321-023-00769-x Engenheiros biomédicos da Duke University desenvolveram uma plataforma de IA que compara moléculas de forma autônoma e aprende com suas variações para antecipar diferenças de propriedades críticas para a descoberta de novos produtos farmacêuticos. A plataforma fornece aos pesquisadores uma ferramenta mais precisa e eficiente para ajudar a projetar produtos terapêuticos e outros produtos químicos com propriedades úteis.
A pesquisa foi publicada em 27 de outubro no Journal of Cheminformatics .
Algoritmos de aprendizado de máquina são cada vez mais usados para estudar e prever as propriedades biológicas, químicas e físicas de pequenas moléculas usadas no desenvolvimento de medicamentos e outras tarefas de design de materiais. Essas ferramentas podem ajudar os pesquisadores a compreender as principais propriedades “ADMET” de uma molécula – como ela é absorvida, distribuída, metabolizada, excretada e sua toxicidade dentro do corpo. Ao compreender estas diferentes propriedades, os investigadores podem identificar moléculas para desenvolver novas terapêuticas mais seguras e eficazes.
Embora as plataformas de aprendizado de máquina existentes permitam aos pesquisadores rastrear um número muito maior de moléculas do que seria possível fabricando-as fisicamente em um laboratório, elas só podem prever as propriedades de uma molécula por vez, limitando sua eficiência geral quando encarregadas de identificar o composto mais ideal.
Embora existam algumas outras abordagens computacionais para eliminar essa etapa extra e comparar diretamente as moléculas, elas são limitadas em seu escopo. Por exemplo, métodos como perturbação de energia livre são muito precisos, mas tão complexos computacionalmente que só podem avaliar um punhado de moléculas por vez. Abordagens como pares moleculares combinados, por outro lado, são muito mais rápidas, mas só podem comparar moléculas muito semelhantes, limitando a sua utilização mais ampla.
Para resolver esse problema, Reker e Zachary Fralish, Ph.D. estudante do laboratório Reker, desenvolveu DeepDelta, uma abordagem de aprendizado profundo que pode comparar eficientemente duas moléculas simultaneamente e prever as diferenças de propriedades entre elas, mesmo que sejam muito diferentes.
“Ao fazer com que a rede aprenda a partir de uma comparação um a um, você está fornecendo mais pontos de dados do que se estivesse aprendendo com uma molécula de cada vez”, disse Reker. “A plataforma está aprendendo sobre a estrutura e as propriedades de cada molécula individualmente, mas também está aprendendo sobre as diferenças entre as duas e como essas diferenças informam as propriedades da molécula”.
A equipe testou a plataforma DeepDelta em dois modelos de última geração na área:Random Forest, um modelo clássico de aprendizado de máquina amplamente utilizado, e ChemProp, uma rede neural profunda na qual o DeepDelta se baseia. Cada sistema comparou duas estruturas moleculares conhecidas e previu 10 propriedades diferentes do ADMET, incluindo como as moléculas são eliminadas dos rins, suas respectivas meias-vidas e quão bem podem ser metabolizadas pelo fígado.
DeepDelta provou ser significativamente mais eficaz e preciso na previsão e quantificação das diferenças nas propriedades moleculares entre moléculas do que as plataformas existentes.
“O treinamento em diferenças moleculares permite que este método seja mais preciso ao decidir se um novo produto químico é melhor ou pior do que o atual”, disse Fralish. "É como fazer o dever de casa mais parecido com o seu teste. Também expandimos bastante o tamanho de nossos conjuntos de dados por meio do emparelhamento, essencialmente dando aos nossos modelos mais lição de casa, o que realmente ajuda as redes neurais que consomem muitos dados a aprender mais."
A equipe está agora ansiosa para incorporar esse modelo em seu trabalho à medida que projeta novas terapias potenciais e otimiza os candidatos a medicamentos existentes.
“Com esta ferramenta, poderíamos analisar um medicamento que quase passou pela aprovação da FDA, mas talvez tivesse problemas de toxicidade hepática, por isso não conseguiu”, disse Fralish. "O DeepDelta pode ajudar a identificar moléculas que tenham as mesmas boas propriedades, mas sem toxicidade hepática. Esta ferramenta abre muitas oportunidades, ajudando-nos a decidir qual produto químico tem a melhor chance de fazer o que queremos no mundo real, economizando tempo e dinheiro. "
Mais informações: Zachary Fralish et al, DeepDelta:prevendo melhorias ADMET de derivados moleculares com aprendizagem profunda, Journal of Cheminformatics (2023). DOI:10.1186/s13321-023-00769-x Fornecido pela Duke University