Simulações moleculares de misturas de amônia apoiam a busca por combustíveis renováveis
 Diferentes misturas de moléculas exibem diferentes propriedades termodinâmicas, que afetam o comportamento das aplicações de engenharia química. Crédito:TU Berlim
Diferentes misturas de moléculas exibem diferentes propriedades termodinâmicas, que afetam o comportamento das aplicações de engenharia química. Crédito:TU Berlim Amônia (NH
3 ) é uma molécula importante com muitas aplicações. Produto final do famoso processo Haber-Bosch, é comumente sintetizado para capturar nitrogênio para fertilizantes e é usado para refrigeração, em produtos de limpeza e na produção de produtos farmacêuticos. Recentemente, esta molécula modesta também atraiu interesse como um recurso potencial para enfrentar um dos desafios mais prementes da atualidade:a necessidade de combustíveis renováveis confiáveis e abundantes.
A amônia é estável e segura de manusear, é combustível e contém a maior fração de hidrogênio de qualquer molécula, exceto o próprio hidrogênio puro. Estes factores prometem torná-lo uma alternativa viável aos vectores energéticos baseados no carbono que estão a impulsionar as alterações climáticas. A pesquisa começou a explorar como a amônia poderia ser usada para alimentar diretamente motores, turbinas a gás e células de combustível de hidrogênio, por exemplo. Acredita-se também que a amônia poderia ser usada para armazenar energia para momentos em que outras energias renováveis, como a energia eólica e a solar, não conseguem atender à demanda.
Muito se sabe sobre a amônia, mas esse interesse em utilizá-la como combustível iniciou uma busca por novas tecnologias de amônia. Isto, por sua vez, levou a uma necessidade crescente entre os engenheiros químicos de dados precisos que descrevem as propriedades termodinâmicas fundamentais da amônia. Essas propriedades incluem uma ampla variedade de características mensuráveis, como equilíbrio de fase, densidade ou capacidade térmica, por exemplo, que caracterizam os sistemas físicos e determinam como funcionam os processos químicos. No caso da amônia, os engenheiros também gostariam de ter um melhor conhecimento de como essas propriedades mudam quando a amônia é misturada com outras moléculas. Esse conhecimento poderia ajudá-los a otimizar processos e condições operacionais.
Jadran Vrabec, atualmente diretor do Instituto de Ciências de Processo da Universidade Técnica de Berlim, passou grande parte de sua carreira usando computação de alto desempenho (HPC) para investigar propriedades termodinâmicas em nível molecular. “As propriedades termodinâmicas são 100% determinadas pelas interações moleculares”, explica. “E como essas interações acontecem tão rápido e em escala tão pequena, só é possível estudá-las realizando grandes simulações usando supercomputadores”.
Em um artigo recente publicado no Journal of Chemical &Engineering Data , ele e o coautor Erich Mace, da TU Berlin, relatam os resultados de simulações focadas nas propriedades termodinâmicas de misturas contendo amônia. Produzidos usando o supercomputador Hawk no Centro de Computação de Alto Desempenho de Stuttgart (HLRS), seus resultados agregam dados valiosos que poderiam apoiar o desenvolvimento de novas aplicações de amônia. Os resultados também poderão ajudar a avaliar a precisão de outros dados existentes, garantindo que os engenheiros tenham as melhores informações disponíveis para trabalhar com a substância.
 Simulações de propriedades termodinâmicas de misturas de amônia e outras moléculas forneceram insights sobre seu equilíbrio líquido-vapor e constantes de Henry, fatores importantes na determinação de como gases e líquidos se misturarão em processos de engenharia química. Crédito:Journal of Chemical &Engineering Data (2023). DOI:10.1021/acs.jced.3c00327
Simulações de propriedades termodinâmicas de misturas de amônia e outras moléculas forneceram insights sobre seu equilíbrio líquido-vapor e constantes de Henry, fatores importantes na determinação de como gases e líquidos se misturarão em processos de engenharia química. Crédito:Journal of Chemical &Engineering Data (2023). DOI:10.1021/acs.jced.3c00327
Simulações em grande escala fornecem insights exclusivos sobre propriedades termodinâmicas
Vrabec é usuário de longa data de recursos de supercomputação HLRS para dinâmica molecular e simulações de Monte Carlo. Sua abordagem se baseia em conceitos de termodinâmica que foram articulados pela primeira vez por Ludwig Boltzmann no século XIX, mas que só se tornaram práticos para aplicação na década de 1950, com a chegada dos primeiros computadores. Desde então, o campo avançou paralelamente ao desenvolvimento de supercomputadores maiores e mais rápidos, a tal ponto que as simulações de Vrabec rastreiam agora os movimentos individuais e as interações de bilhões ou mesmo trilhões de moléculas simultaneamente. Usando o software desenvolvido por seu laboratório para capturar seletivamente dados de interesse, ele pode então estudar as propriedades termodinâmicas das moléculas.
Vrabec usa dois códigos de simulação chamados ms2 e ls1, que ele desenvolveu e otimizou ao longo de uma longa e frutífera colaboração com os membros da equipe do HLRS, Martin Bernreuther e Christoph Niethammer. Em 2019, a equipe ainda estabeleceu um recorde mundial para o maior sistema molecular já simulado usando métodos de dinâmica molecular. Usando ls1, eles escalaram eficientemente seu código para um sistema de 21 trilhões de átomos no qual cada molécula individual e suas interações com outras moléculas poderiam ser rastreadas.
No trabalho recente sobre amônia, Mace e Vrabec realizaram dinâmica molecular e simulações de Monte Carlo usando ms2 para investigar cinco misturas comumente usadas envolvendo amônia em processos de engenharia química:argônio-amônia, metano-amônia, hidrogênio-amônia, nitrogênio-amônia e oxigênio. -amônia. Para cada mistura, as simulações geraram dados que descrevem o equilíbrio vapor-líquido (VLE) – uma medição da distribuição de moléculas em um sistema através das fases vapor ou líquida – para uma ampla faixa de temperaturas e pressões.
Em seu artigo, Mace e Vrabec apontam que os dados VLE são frequentemente usados no desenvolvimento de equações de estado para fluidos industriais; isto é, os dados podem ser usados para prever o estado da matéria sob diferentes condições físicas devido a mudanças de temperatura, pressão, volume ou composição. Essas informações são essenciais para determinar misturas e condições de trabalho ideais em aplicações industriais.
As simulações moleculares de Vrabec são particularmente valiosas porque podem ser usadas para investigar uma gama muito mais ampla de escalas do que seria possível usando abordagens experimentais.
“Em nossas simulações, fornecemos medições de propriedades termodinâmicas até pressões de 50 megapascais. Isso é 500 vezes a pressão do ar ambiente”, observa Vrabec. "Embora os dados sobre misturas de amônia tenham sido coletados há mais de um século, a cobertura dos dados é surpreendentemente estreita. A razão é que o esforço para medi-los experimentalmente é proibitivamente enorme. Seria necessário equipamento especial caro e perigoso de operar. Em simulações de computador, podemos obter resultados de forma segura e relativamente barata." Seus métodos também fornecem um nível de precisão comparável ao de abordagens experimentais em faixas onde dados experimentais estão disponíveis.
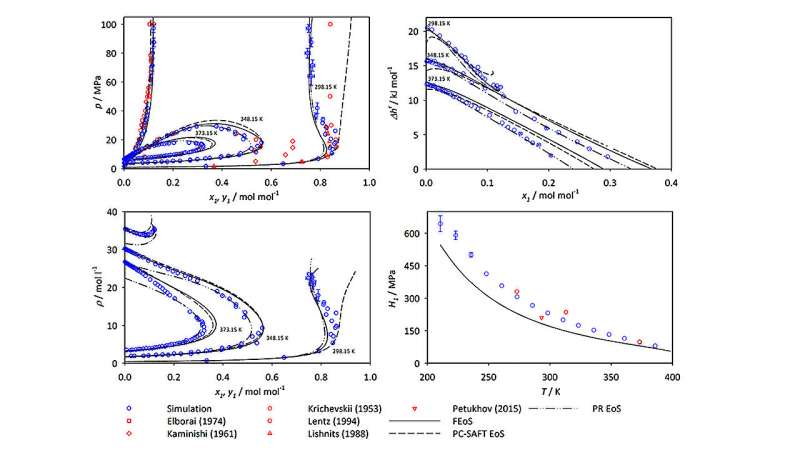 Os gráficos comparam dados de simulação e experimentais para misturas de amônia e metano em uma ampla gama de composições, pressões e temperaturas. Os dados de simulação (representados em círculos azuis) correspondem bem a outros dados experimentais e revelam valores discrepantes em dados experimentais (vistos, por exemplo, nos losangos vermelhos dos resultados de Kaminishi em 1961, na metade inferior da figura superior esquerda) que provavelmente ser impreciso. Crédito:Journal of Chemical &Engineering Data (2023). DOI:10.1021/acs.jced.3c00327
Os gráficos comparam dados de simulação e experimentais para misturas de amônia e metano em uma ampla gama de composições, pressões e temperaturas. Os dados de simulação (representados em círculos azuis) correspondem bem a outros dados experimentais e revelam valores discrepantes em dados experimentais (vistos, por exemplo, nos losangos vermelhos dos resultados de Kaminishi em 1961, na metade inferior da figura superior esquerda) que provavelmente ser impreciso. Crédito:Journal of Chemical &Engineering Data (2023). DOI:10.1021/acs.jced.3c00327
Melhores dados para pesquisa de amônia
Quando Mace e Vrabec analisaram seus dados de simulação, mostraram que, embora a amônia seja um componente em todos os cinco sistemas que estudaram, os gráficos resultantes dos valores de VLE parecem dramaticamente diferentes para diferentes misturas moleculares. De acordo com Vrabec, “O comportamento de fase de diferentes misturas é fortemente determinado pelas interações entre as moléculas do sistema. Você precisa entender essas propriedades se estiver interessado em trabalhar com misturas de amônia”.
O artigo e seus dados suplementares oferecem mais de 400 novos pontos de dados para cada mistura estudada. Usando o Hawk, eles foram capazes de produzir os resultados de cada mistura em apenas alguns dias de computação. Os resultados serão de particular valor para condições extremas e difíceis de estudar, para as quais há poucos dados disponíveis, e poderão ajudar os engenheiros a identificar pontos ideais onde as condições seriam ideais para o processamento eficiente de amônia.
O estudo incluiu novos dados de simulação e dados publicados anteriormente na literatura científica, permitindo que Mace e Vrabec comparassem seus resultados com outros conjuntos de dados existentes de valores de VLE. Na maioria das situações, os seus resultados corresponderam estreitamente aos de estudos anteriores. Em alguns casos, no entanto, identificaram divergências significativas entre os seus resultados e as medições e previsões geradas experimentalmente por outros grupos de investigação. Os autores atribuem essas discrepâncias a limitações ou imprecisões nos métodos experimentais correspondentes. Eles também sugerem que fontes de dados experimentais específicas devem ser usadas com cautela em pesquisas futuras ou aplicações de engenharia química.
Vrabec diz que em trabalhos recentes ele se concentrou principalmente na simulação de propriedades termodinâmicas de sistemas moleculares, geralmente na escala submicrométrica. Apesar das muitas ordens de grandeza que se situam entre esta escala e o nível dos processos observáveis, existem métodos precisos para traduzir estes conhecimentos a nível molecular em previsões úteis do mundo real.
À medida que os supercomputadores crescem, no entanto, ele prevê que também poderá ser possível simular não apenas propriedades, mas também processos termodinâmicos usando condições de contorno próximas às aplicações do mundo real. O aumento do desempenho do HPC poderia produzir resultados mais precisos sobre fenômenos dinâmicos com uma melhor relação sinal-ruído.
Enquanto isso, porém, os resultados de sua equipe demonstram o valor da dinâmica molecular e da simulação de Monte Carlo usando computação de alto desempenho, e fornecerão uma nova compreensão do comportamento das fases que os engenheiros podem usar para desenvolver novas tecnologias baseadas em amônia.