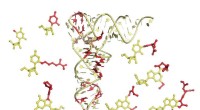
Descobrir o que faz algumas proteínas brilharem requer uma compreensão da química. Crédito:eLife - a revista, CC BY-SA
A inteligência artificial mudou a forma como a ciência é feita, permitindo que os pesquisadores analisem as enormes quantidades de dados que os instrumentos científicos modernos geram. Ele pode encontrar uma agulha em um milhão de pilhas de informações e, usando aprendizado profundo, pode aprender com os próprios dados. A IA está acelerando os avanços na busca de genes, medicina, design de medicamentos e criação de compostos orgânicos.
O aprendizado profundo usa algoritmos, geralmente redes neurais treinadas em grandes quantidades de dados, para extrair informações de novos dados. É muito diferente da computação tradicional com suas instruções passo a passo. Em vez disso, ele aprende com os dados. O aprendizado profundo é muito menos transparente do que a programação de computador tradicional, deixando questões importantes – o que o sistema aprendeu, o que ele sabe?
Como professor de química, gosto de projetar testes que tenham pelo menos uma pergunta difícil que expanda o conhecimento dos alunos para estabelecer se eles podem combinar ideias diferentes e sintetizar novas ideias e conceitos. Criamos essa pergunta para o garoto-propaganda dos defensores da IA, AlphaFold, que resolveu o problema de dobramento de proteínas.
Dobramento de proteínas As proteínas estão presentes em todos os organismos vivos. Eles fornecem estrutura às células, catalisam reações, transportam pequenas moléculas, digerem alimentos e fazem muito mais. Eles são compostos de longas cadeias de aminoácidos como contas em uma corda. Mas para que uma proteína faça seu trabalho na célula, ela deve torcer e dobrar em uma estrutura tridimensional complexa, um processo chamado de dobramento de proteínas. Proteínas mal dobradas podem levar a doenças.
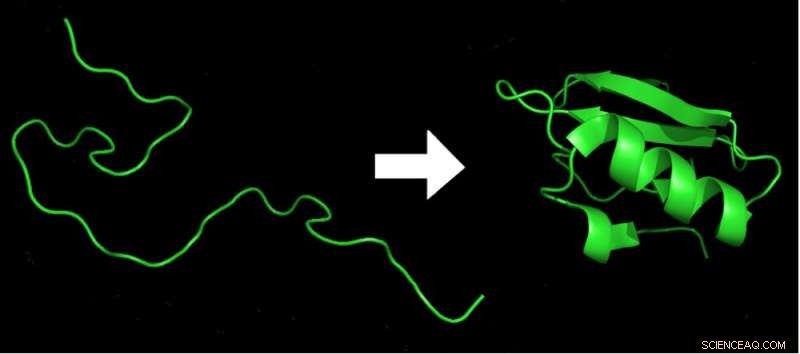
Dentro de milissegundos da saída de uma cadeia de aminoácidos (esquerda) do ribossomo, ela é dobrada na forma 3D de menor energia (direita), que é necessária para a função da proteína. Crédito:Marc Zimmer, CC BY-ND
Em seu discurso de aceitação do Nobel de química em 1972, Christiaan Anfinsen postulou que deveria ser possível calcular a estrutura tridimensional de uma proteína a partir da sequência de seus blocos de construção, os aminoácidos.
Assim como a ordem e o espaçamento das letras neste artigo dão sentido e mensagem, a ordem dos aminoácidos determina a identidade e a forma da proteína, o que resulta em sua função.
Devido à flexibilidade inerente dos blocos de construção de aminoácidos, uma proteína típica pode adotar cerca de 10 elevado a 300 formas diferentes. Este é um número enorme, mais do que o número de átomos no universo. No entanto, em um milissegundo, cada proteína em um organismo se dobrará em sua própria forma específica - o arranjo de energia mais baixa de todas as ligações químicas que compõem a proteína. Mude apenas um aminoácido nas centenas de aminoácidos normalmente encontrados em uma proteína e ela pode se dobrar e não funcionar mais.
AlphaFold Por 50 anos, cientistas da computação tentaram resolver o problema de dobramento de proteínas – com pouco sucesso. Então, em 2016, a DeepMind, uma subsidiária de IA da Alphabet, controladora do Google, iniciou seu programa AlphaFold. Ele usou o banco de dados de proteínas como seu conjunto de treinamento, que contém as estruturas determinadas experimentalmente de mais de 150.000 proteínas.
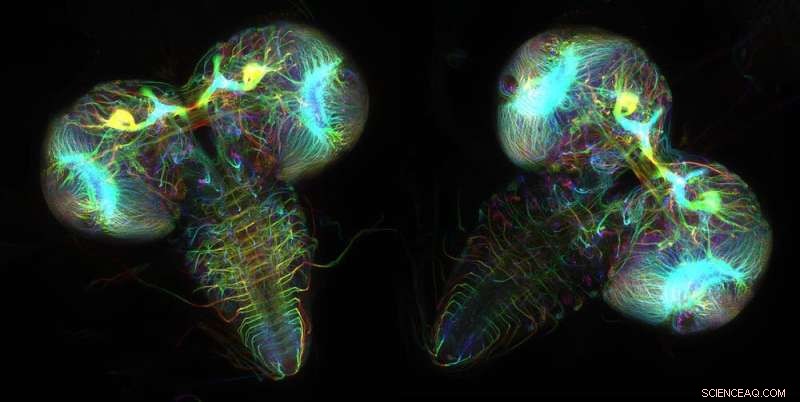
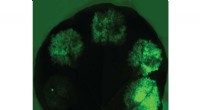
Neurônios que expressam proteínas fluorescentes revelam as estruturas cerebrais de duas larvas de moscas-das-frutas. Crédito:Wen Lu e Vladimir I. Gelfand, Feinberg School of Medicine, Northwestern University
Em menos de cinco anos, o AlphaFold venceu o problema de dobramento de proteínas – pelo menos a parte mais útil dele, ou seja, determinar a estrutura da proteína a partir de sua sequência de aminoácidos. AlphaFold não explica como as proteínas se dobram com tanta rapidez e precisão. Foi uma grande vitória para a IA, porque não apenas acumulou enorme prestígio científico, mas também foi um grande avanço científico que poderia afetar a vida de todos.
Hoje, graças a programas como AlphaFold2 e RoseTTAFold, pesquisadores como eu podem determinar a estrutura tridimensional das proteínas a partir da sequência de aminoácidos que compõem a proteína – sem nenhum custo – em uma ou duas horas. Antes do AlphaFold2, tínhamos que cristalizar as proteínas e resolver as estruturas usando cristalografia de raios X, um processo que levava meses e custava dezenas de milhares de dólares por estrutura.
Agora também temos acesso ao AlphaFold Protein Structure Database, onde a Deepmind depositou as estruturas 3D de quase todas as proteínas encontradas em humanos, camundongos e mais de 20 outras espécies. Até o momento, eles resolveram mais de um milhão de estruturas e planejam adicionar outras 100 milhões de estruturas somente este ano. O conhecimento das proteínas disparou. A estrutura de metade de todas as proteínas conhecidas provavelmente será documentada até o final de 2022, entre elas muitas novas estruturas únicas associadas a novas funções úteis.
Pensando como um químico O AlphaFold2 não foi projetado para prever como as proteínas interagem umas com as outras, mas foi capaz de modelar como as proteínas individuais se combinam para formar grandes unidades complexas compostas por várias proteínas. Tínhamos uma pergunta desafiadora para o AlphaFold - seu conjunto de treinamento estrutural ensinou alguma química? Poderia dizer se os aminoácidos reagiriam uns com os outros - uma ocorrência rara, mas importante?
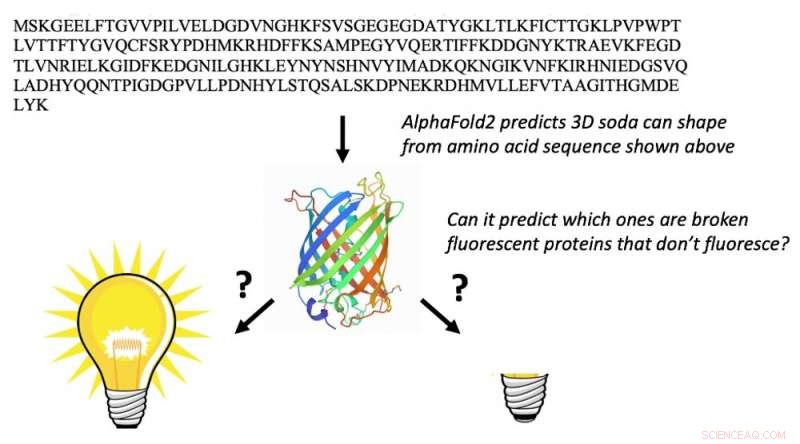
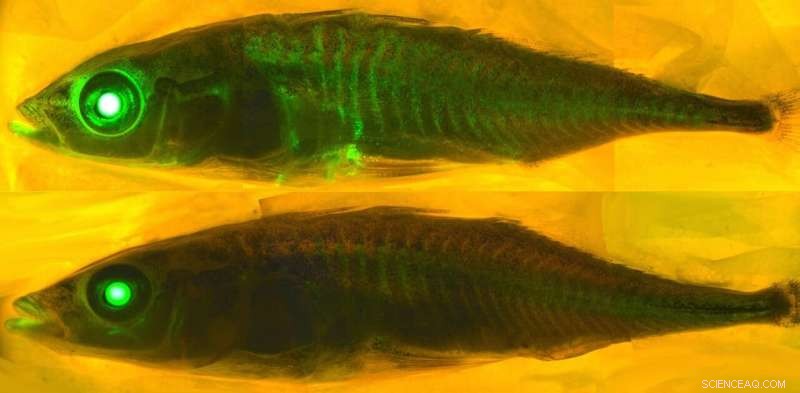
AlphaFold2 pode pegar a sequência de aminoácidos de proteínas fluorescentes (letras no topo) e prever suas formas de barril 3D (no meio). Isso não é surpreendente. O que é totalmente inesperado é que também pode prever quais proteínas fluorescentes estão “quebradas” e não podem fluorescer. Crédito:Marc Zimmer, CC BY-ND
Eu sou um químico computacional interessado em proteínas fluorescentes. Estas são proteínas encontradas em centenas de organismos marinhos como águas-vivas e corais. Seu brilho pode ser usado para iluminar e estudar doenças.
Existem 578 proteínas fluorescentes no banco de dados de proteínas, das quais 10 estão "quebradas" e não fluorescem. As proteínas raramente atacam a si mesmas, um processo chamado modificação pós-tradução autocatalítica, e é muito difícil prever quais proteínas reagirão consigo mesmas e quais não reagirão.
Apenas um químico com uma quantidade significativa de conhecimento de proteínas fluorescentes seria capaz de usar a sequência de aminoácidos para encontrar as proteínas fluorescentes que têm a sequência de aminoácidos correta para sofrer as transformações químicas necessárias para torná-las fluorescentes. Quando apresentamos o AlphaFold2 com as sequências de 44 proteínas fluorescentes que não estão no banco de dados de proteínas, ele dobrou as proteínas fluorescentes fixas de forma diferente das quebradas.
O resultado nos surpreendeu:AlphaFold2 aprendeu um pouco de química. Descobriu quais aminoácidos nas proteínas fluorescentes fazem a química que as faz brilhar. Suspeitamos que o conjunto de treinamento do banco de dados de proteínas e os alinhamentos de sequências múltiplas permitem que o AlphaFold2 "pense" como os químicos e procure os aminoácidos necessários para reagir uns com os outros para tornar a proteína fluorescente.
Um programa de dobradura aprendendo alguma química de seu conjunto de treinamento também tem implicações mais amplas. Ao fazer as perguntas certas, o que mais pode ser obtido com outros algoritmos de aprendizado profundo? Os algoritmos de reconhecimento facial poderiam encontrar marcadores ocultos para doenças? Poderiam algoritmos projetados para prever padrões de gastos entre os consumidores também encontrar uma propensão para pequenos roubos ou fraudes? E o mais importante, essa capacidade – e saltos semelhantes em capacidade em outros sistemas de IA – é desejável?
+ Explorar mais As proteínas lembram?
Este artigo é republicado de The Conversation sob uma licença Creative Commons. Leia o artigo original.