A rede neural detecta locais de ligação de proteína-peptídeo para iniciar a descoberta de peptídeo de drogas
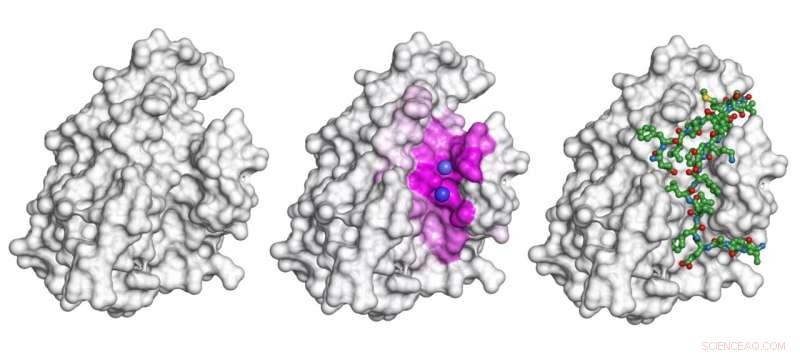 p A forma cinza é uma proteína. Para o cenário de ligação desta proteína ao peptídeo mostrado como um modelo esverdeado em forma de bastão e bola à direita, o modelo apresentado no estudo destaca a superfície envolvida na interação (a área rosa no meio) e prevê os locais exatos de ligação (esferas roxas). Crédito:Igor Kozlovskii e Petr Popov / Skoltech
p A forma cinza é uma proteína. Para o cenário de ligação desta proteína ao peptídeo mostrado como um modelo esverdeado em forma de bastão e bola à direita, o modelo apresentado no estudo destaca a superfície envolvida na interação (a área rosa no meio) e prevê os locais exatos de ligação (esferas roxas). Crédito:Igor Kozlovskii e Petr Popov / Skoltech
p Dois pesquisadores da Skoltech apresentaram um modelo de rede neural altamente eficiente que usa dados sobre a estrutura das proteínas para prever quais de suas partes interagem com outras moléculas biológicas chamadas de peptídeos. Saber disso é útil para o desenvolvimento de drogas com base em peptídeos, que pode afetar as interações proteína-proteína dentro das células de uma forma direcionada e não tóxica, regulando uma ampla gama de processos celulares. O estudo saiu no
Journal of Chemical Information and Modeling . p As proteínas são a maquinaria das células, movendo-se, engajando-se uns com os outros, e executando todos os tipos de operações. Os farmacologistas sempre ficaram intrigados com a perspectiva de mexer nas interações entre as proteínas. No entanto, eles pareciam estar fora dos limites como um alvo potencial de drogas:as moléculas terapêuticas maiores, chamados biológicos, não conseguiu penetrar na célula para agir sobre as proteínas, enquanto os agentes de pequenas moléculas freqüentemente se mostraram incapazes de tal ação.
p Peptides, que naturalmente medeiam ou regulam cerca de 40% dos processos celulares, ocupar um meio-termo promissor e manter perspectivas para medicamentos visando interações proteína-proteína. Os peptídeos oferecem o melhor dos dois mundos:como pequenas moléculas, eles podem penetrar na membrana celular para realmente alcançar seus alvos, e também exibem baixa toxicidade, junto com alta afinidade e especificidade (ação forte e focada) - as marcas registradas dos produtos biológicos.
p Para projetar drogas baseadas em peptídeos, os farmacologistas precisam conhecer os chamados locais de ligação para qualquer proteína-alvo. Isso é, os pontos na proteína que podem se ligar a um peptídeo. Quanto mais esses sites são conhecidos, quanto mais oportunidades para o desenho de medicamentos estiverem disponíveis.
p Os pesquisadores podem identificar os locais de ligação experimentalmente, por exemplo, usando cristalografia de raios-X, que revela a estrutura 3D das proteínas cristalizadas, estudando como elas difratam os raios-X. Mas isso é muito caro para uma longa lista de moléculas, e os métodos computacionais oferecem uma alternativa mais rápida e barata. Alguns deles usam técnicas de aprendizado de máquina, e à medida que mais dados sobre as estruturas dos complexos proteína-peptídeo são acumulados, esses métodos se tornam mais poderosos e fornecem previsões de sites de ligação cada vez melhores.
p Em seu artigo de 22 de julho no
Journal of Chemical Information and Modeling , Skoltech Ph.D. o aluno Igor Kozlovskii e o professor assistente Petr Popov do grupo iMolecule apresentaram um método computacional chamado BiteNetPp, que aproveita o poder das redes neurais convolucionais 3D para detectar locais de ligação de proteína-peptídeo. No BiteNetPp, uma estrutura de proteína conhecida é alimentada a uma rede neural, que então destaca os locais de ligação de peptídeo suspeitos, e produz um conjunto de coordenadas 3D putativas, junto com as pontuações de probabilidade associadas.
p Petr Popov comenta sobre a abordagem de detecção de sites de ligação como reconhecimento de imagem, originalmente introduzido no artigo anterior da equipe e transportado para o estudo relatado nesta história:"Assim como as redes neurais podem ser treinadas para reconhecer, dizer, pedestres ou ciclistas em fotos 2D comuns, vemos a detecção de sites de ligação como a localização de um tipo específico de objeto em uma imagem. A diferença é que usamos dados de estrutura atômica 3D como nossas entradas, então o modelo opera em 'voxels, "um análogo tridimensional de pixels."
p O modelo recém-apresentado, na verdade, baseia-se no modelo do artigo anterior. "Isso é chamado de adaptação de domínio. BiteNetPp é o primeiro modelo a ter sido ajustado em um conjunto de dados de proteína-peptídeo após ser inicialmente treinado em dados de moléculas pequenas de proteína, "Popov explica." Você pode imaginar isso como o treinamento de um modelo para identificar lugares onde os ciclistas tendem a parar na rua, mas você começa com dados sobre onde os pedestres tendem a parar - e só então estende seu domínio aos ciclistas. Em vez de começar do zero, você retreina o modelo, antecipando que os 'locais de ligação' para os ciclistas podem ter algumas semelhanças com aqueles que atraem os pedestres:você sabe, carrinhos de sorvete, luzes de trânsito, esse tipo de coisas."
p Os criadores do modelo demonstraram que o BiteNetPp supera consistentemente os métodos de última geração existentes, comparando suas previsões para os sítios de ligação de proteína-peptídeo que são conhecidos por meio de observações experimentais. Mais importante, o novo modelo leva menos de um segundo para analisar a estrutura de uma única proteína, tornando-o adequado para estudos em grande escala. Existem milhares de interações proteína-proteína potencialmente alvejáveis por drogas baseadas em peptídeos, portanto, os métodos computacionais devem ser rápidos o suficiente para tornar sua triagem viável em um contexto farmacológico.