Crédito CC0:domínio público
Muitos produtos farmacêuticos atuam visando o que é conhecido como 'receptores acoplados à proteína G'. Em um novo estudo, Cientistas da Universidade de Uppsala descrevem como foram capazes de prever como moléculas especiais que podem ser usadas em uma nova imunoterapia contra o câncer se ligam a esses receptores. Os métodos de cálculo dos pesquisadores, apresentado no jornal Angewandte Chemie são uma contribuição vital para o futuro design de medicamentos com base na estrutura.
Os receptores acoplados à proteína G (GPCRs) estão entre os grupos-alvo de proteínas de maior importância para o desenvolvimento de medicamentos. Esses receptores reagem a, por exemplo, luz, sabores, cheiros, adrenalina, histamina, dopamina e uma longa lista de outras moléculas, transmitindo mais sinais bioquímicos dentro das células. Os pesquisadores que realizaram o levantamento dos GPCRs foram agraciados com o Prêmio Nobel de Química em 2012.
Hoje, cerca de 30 por cento de todos os medicamentos no mercado têm GPCRs como proteínas-alvo. Algumas moléculas de drogas, como morfina, ativam os receptores (agonistas) enquanto outros, como bloqueadores beta, inativá-los (antagonistas).
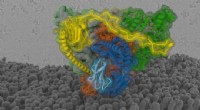
Um GPCR importante é o receptor de adenosina A2A. Seus antagonistas podem ser usados em nova imunoterapia contra o câncer. Em conjunto com a empresa biofarmacêutica Sosei-Heptares, os pesquisadores Willem Jespers, Johan Åqvist e Hugo Gutierrez-de-Terán, da Universidade de Uppsala, conseguiram mostrar como uma série de antagonistas A2A se ligam ao receptor e o inativam.
Com simulações de dinâmica molecular e cálculo de energias de ligação, tornou-se possível prever como as moléculas da empresa farmacêutica se ligariam aos receptores e com que força o fazem. Depois disso, novos antagonistas foram projetados, e sintetizado por químicos da Universidade de Santiago de Compostela, Espanha. Estruturas tridimensionais dos complexos que se formam entre essas moléculas e o receptor foram então determinadas experimentalmente com cristalografia de raios-X. Os cálculos do computador provaram ser capazes de prever a estrutura e a força de ligação nos complexos com alta precisão.
"Este é um passo sólido à frente, e conseguimos prever com grande precisão como essa família de moléculas se liga ao receptor A2A. Nossos métodos de cálculo estão agora tendo um grande avanço no design de medicamentos com base na estrutura, "diz Hugo Gutierrez-de-Terán, que liderou o projeto do grupo de Uppsala.