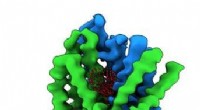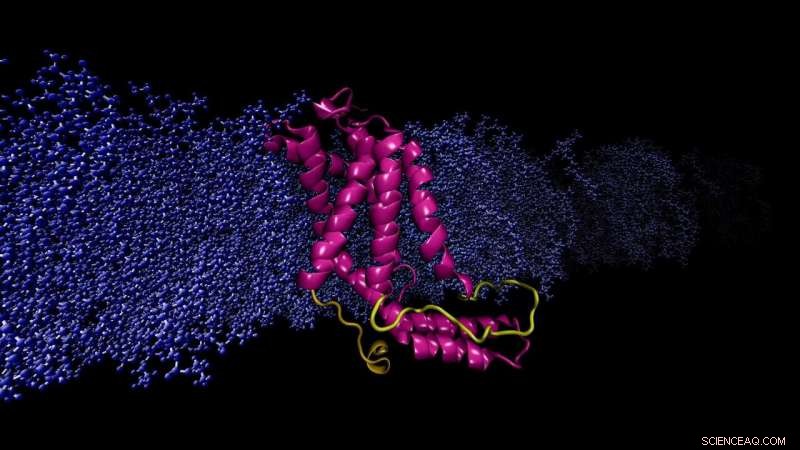
Modelo computacional de membrana celular embutida na proteína YidC2. O loop modelado (amarelo), faltando na estrutura de cristal de raios-x, é crucial para a estabilização da proteína. Crédito:Sogol Moradi
Um novo estudo feito por químicos da Universidade de Arkansas mostra que a cristalografia de raios-X, o método padrão para determinar a estrutura das proteínas, pode fornecer informações imprecisas sobre um conjunto crítico de proteínas - aquelas encontradas nas membranas celulares - que, por sua vez, podem estar levando a um design de drogas pobre e ineficiente.
As descobertas dos pesquisadores foram publicadas hoje em Relatórios Científicos , uma publicação da Nature.
"Dois terços de todas as drogas, incluindo aqueles usados para quimioterapia, proteínas alvo encontradas nas membranas celulares, "disse Mahmoud Moradi, professor assistente de química e bioquímica no J. William Fulbright College of Arts and Sciences. "Infelizmente, Cristalografia de raio-x, o padrão ouro para determinar a estrutura das proteínas, tem muitas limitações ao lidar com aquelas encontradas na membrana celular. Nosso trabalho expõe, e de muitas maneiras, explica essas limitações. "
Consideradas as moléculas burras de carga das células, as proteínas são responsáveis por quase todas as tarefas nos sistemas vivos. Algumas proteínas vivem dentro das células, e alguns residem na membrana da célula, uma camada externa de lipídios que separa a célula de seu ambiente externo. As proteínas da membrana são extremamente importantes porque regulam a troca de informações e materiais entre a célula e seu ambiente, uma tarefa vital para a sobrevivência e função normal da célula porque qualquer distúrbio na função da proteína pode resultar em doença.
O estudo da função da proteína é necessário para a compreensão da base molecular da doença. Para fazer isso, pesquisadores têm contado com cristalografia de raios-X, a principal ferramenta para determinar a forma e a estrutura das proteínas. A cristalografia de raios-X também é essencial para o propósito de projetar drogas que manipulem com eficiência a função das proteínas. Contudo, o estudo da estrutura da proteína da membrana é difícil porque seu ambiente nativo não é compatível com a cristalografia de raios-X. Os pesquisadores devem remover as proteínas de seu ambiente nativo e colocá-las em um ambiente lipídico artificial antes de aplicar a técnica.
Moradi e Thomas Harkey - um estudante de graduação na época e agora um estudante de medicina na Universidade de Arkansas para Ciências Médicas - abordaram esse problema de um ângulo diferente. Por cerca de dois anos, eles usaram um supercomputador no Arkansas High Performance Computing Center para funcionar continuamente, cálculos de nível de microssegundo simulando a dinâmica molecular de YidC2, uma proteína de membrana com uma alça citoplasmática não resolvida cristalograficamente em sua estrutura molecular. Os loops citoplasmáticos são conhecidos por terem significado funcional nas proteínas de membrana.
As simulações de Moradi e Harkey demonstraram que o loop citoplasmático de YidC2 estabilizou a proteína inteira, particularmente a região C1, uma área potencialmente importante para o desenho de medicamentos. Grupos de cabeça de lipídios altamente polares ou carregados interagiram e estabilizaram a alça. Este achado demonstrou que loops não resolvidos de proteínas de membrana podem ser importantes para a estabilização de proteínas, apesar da aparente falta de estrutura molecular.
"Tipicamente, se parte de uma proteína não for resolvida na cristalografia de raios-X, é interpretado como carente de uma estrutura particular, "Disse Moradi." Nós mostramos que para as proteínas de membrana e, particularmente, partes da proteína que interagem com a membrana celular, esta interpretação não é precisa e pode ser enganosa. Achamos que a explicação alternativa para o distúrbio pode ser que a proteína não é estudada em seu ambiente de membrana nativa. "
Moradi disse que seus resultados também demonstraram que a química computacional e a tecnologia de supercomputação podem ser usadas para modelar proteínas de membrana com mais precisão em um ambiente que imita seu ambiente fisiológico.