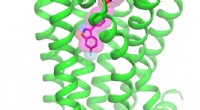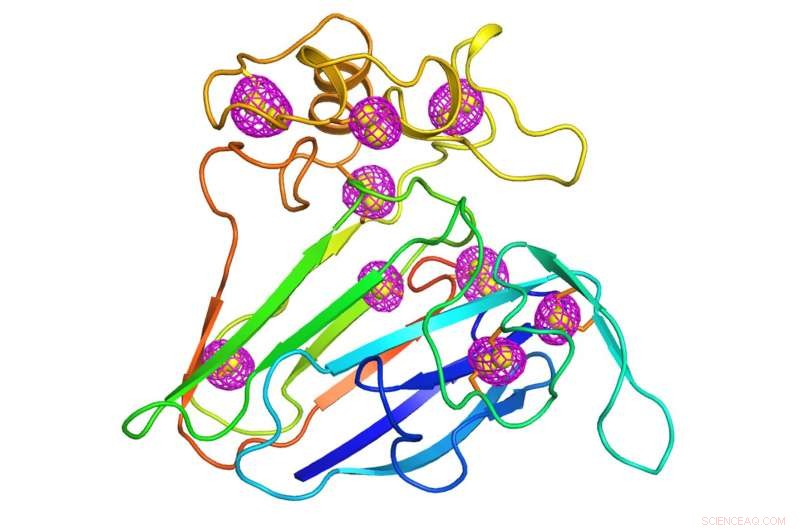
Um desenho animado que representa a estrutura de uma proteína vegetal bem estudada que serviu de caso de teste para a recém-desenvolvida técnica de microcristalografia. Padrões de malha magenta em torno dos átomos de enxofre intrínsecos à proteína (esferas amarelas) indicam os sinais anômalos que foram extraídos usando difração de raios-X de baixa energia de milhares de cristais medindo menos de 10 milionésimos de metro, do tamanho de uma bactéria. Crédito:Laboratório Nacional de Brookhaven
O uso de raios-x para revelar as estruturas 3-D em escala atômica das proteínas levou a incontáveis avanços na compreensão de como essas moléculas funcionam nas bactérias, vírus, plantas, e humanos - e tem orientado o desenvolvimento de medicamentos de precisão para combater doenças como câncer e AIDS. Mas muitas proteínas não podem ser transformadas em cristais grandes o suficiente para que seus arranjos atômicos sejam decifrados. Para enfrentar este desafio, cientistas do Laboratório Nacional Brookhaven do Departamento de Energia dos Estados Unidos (DOE) e colegas da Universidade de Columbia desenvolveram uma nova abordagem para resolver estruturas de proteínas de cristais minúsculos.
O método depende do manuseio de amostra exclusivo, extração de sinal, e abordagens de montagem de dados, e uma linha de luz capaz de focalizar raios-x intensos na Fonte de Luz Síncrotron Nacional II de Brookhaven (NSLS-II) - uma instalação do usuário do DOE Office of Science - em um ponto milionésimo de metro, cerca de um quinquagésimo da largura de um cabelo humano.
"Nossa técnica realmente abre a porta para lidar com microcristais que antes eram inacessíveis, incluindo receptores de superfície celular difíceis de cristalizar e outras proteínas de membrana, proteínas flexíveis, e muitas proteínas humanas complexas, "disse o cientista do Brookhaven Lab, Qun Liu, o autor correspondente no estudo, que foi publicado em 3 de maio, 2019, no IUCrJ , um jornal da União Internacional de Cristalografia.
Decifrando estruturas de proteínas
A cristalografia de proteínas tem sido um método dominante para resolver estruturas de proteínas desde 1958, melhorando ao longo do tempo à medida que as fontes de raios-X se tornam mais poderosas, permitindo determinações de estrutura mais precisas. Para determinar a estrutura de uma proteína, os cientistas medem como os raios-x, como os gerados no NSLS-II, difratam, ou quicar, os átomos em uma rede cristalina ordenada consistindo em muitas cópias da mesma molécula de proteína, todas dispostas da mesma maneira. O padrão de difração transmite informações sobre onde os átomos estão localizados. Mas não é suficiente.
"Apenas as amplitudes das 'ondas' de raios-X difratadas são registradas no detector, mas não suas fases (o tempo entre as ondas), "disse Liu." Ambos são necessários para reconstruir uma estrutura 3-D. Este é o chamado problema de fase cristalográfica. "
Cristalógrafos resolveram esse problema coletando dados de fase de um tipo diferente de espalhamento, conhecido como espalhamento anômalo. O espalhamento anômalo ocorre quando átomos mais pesados do que os principais componentes de carbono de uma proteína, hidrogênio, e o nitrogênio absorve e reemite alguns dos raios-x. Isso acontece quando a energia dos raios X está próxima da energia que esses átomos pesados gostam de absorver. Os cientistas às vezes inserem artificialmente átomos pesados, como selênio ou platina, na proteína para essa finalidade. Mas átomos de enxofre, que aparecem naturalmente em todas as moléculas de proteína, também pode produzir esses sinais, embora mais fraco. Mesmo que esses sinais anômalos sejam fracos, um grande cristal geralmente tem cópias suficientes da proteína com átomos de enxofre suficientes para torná-los mensuráveis. Isso dá aos cientistas as informações de fase necessárias para localizar os átomos de enxofre e traduzir os padrões de difração em uma estrutura 3D completa.
"Depois de saber as posições de enxofre, você pode calcular as fases para os outros átomos de proteína porque a relação entre o enxofre e os outros átomos é fixa, "disse Liu.
Mas pequenos cristais, por definição, não tem tantas cópias da proteína de interesse. Então, em vez de procurar informações de difração e fase de cópias repetidas de uma proteína em um único grande cristal, a equipe de Brookhaven / Columbia desenvolveu uma maneira de fazer medições de muitos cristais minúsculos, e, em seguida, reúna os dados coletivos.
Cristais minúsculos, grandes resultados
Para lidar com os pequenos cristais, a equipe desenvolveu grades de amostra padronizadas com micro poços. Depois de derramar solvente contendo os microcristais sobre essas grades de montagem, os cientistas removeram o solvente e congelaram os cristais que estavam presos nas grades.
“Ainda temos um desafio, no entanto, porque não podemos ver onde os minúsculos cristais estão em nossa grade, "disse Liu." Para descobrir, usamos microdifração na linha de luz de Cristalografia Macromolecular Microfocagem de Fronteira do NSLS-II (FMX) para fazer o levantamento de toda a grade. Escaneando linha por linha, podemos descobrir onde esses cristais estão escondidos. "
Como Martin Fuchs, o cientista-chefe da linha de luz da FMX, explicado, "A linha de luz FMX pode focar a intensidade total do feixe de raios-X até o tamanho de um mícron, ou milionésimo de um metro. Podemos controlar com precisão o tamanho do feixe para combiná-lo com o tamanho dos cristais - cinco mícrons no caso do experimento atual. Esses recursos são cruciais para obter o melhor sinal, " ele disse.
Wuxian Shi, outro cientista da linha de luz FMX, observou que "os dados coletados na pesquisa da grade contêm informações sobre a localização dos cristais. Além disso, também podemos ver o quão bem cada cristal difrata, o que nos permite escolher apenas os melhores cristais para a coleta de dados. "
Os cientistas foram então capazes de manobrar o porta-amostra para colocar cada microcristal mapeado de interesse de volta no centro do feixe de raios-X de precisão para a coleta de dados.
Eles usaram a energia mais baixa disponível na linha de luz - ajustada para se aproximar o mais possível da energia de absorção dos átomos de enxofre - e coletaram dados de espalhamento anômalo.
"A maioria das linhas de luz cristalográficas não conseguiu alcançar a borda de absorção de enxofre para sinais anômalos otimizados, "disse o co-autor Wayne Hendrickson, da Universidade de Columbia." Felizmente, NSLS-II é uma fonte de luz síncrotron líder mundial que fornece raios-X brilhantes cobrindo um amplo espectro de energia de raios-X. E embora nosso nível de energia estivesse ligeiramente acima da energia de absorção ideal para o enxofre, gerou os sinais anômalos de que precisávamos. "
Mas os cientistas ainda tinham algum trabalho a fazer para extrair esses sinais importantes e reunir os dados de muitos cristais minúsculos.
"Na verdade, estamos obtendo milhares de dados, "disse Liu." Usamos cerca de 1400 microcristais, cada um com seu próprio conjunto de dados. Temos que colocar todos os dados desses microcristais juntos. "
They also had to weed out data from crystals that were damaged by the intense x-rays or had slight variations in atomic arrangements.
"A single microcrystal does not diffract x-rays sufficiently for structure solution prior to being damaged by the x-rays, " said Sean McSweeney, deputy photon division director and program manager of the Structural Biology Program at NSLS-II. "This is particularly true with crystals of only a few microns, the size of about a bacterial cell. We needed a way to account for that damage and crystal structure variability so it wouldn't skew our results."
They accomplished these goals with a sophisticated multi-step workflow process that sifted through the data, discarded outliers that might have been caused by radiation damage or incompatible crystals, and ultimately extracted the anomalous scattering signals.
"This is a critical step, " said Liu. "We developed a computing procedure to assure that only compatible data were merged in a way to align the individual microcrystals from diffraction patterns. That gave us the required signal-to-noise ratios for structure determination."
Applying the technique
This technique can be used to determine the structure of any protein that has proven hard to crystallize to a large size. These include cell-surface receptors that allow cells of advanced lifeforms such as animals and plants to sense and respond to the environment around them by releasing hormones, transmitting nerve signals, or secreting compounds associated with cell growth and immunity.
"To adapt to the environment through evolution, these proteins are malleable and have lots of non-uniform modifications, " said Liu. "It's hard to get a lot of repeat copies in a crystal because they don't pack well."
Em humanos, receptors are common targets for drugs, so having knowledge of their varied structures could help guide the development of new, more targeted pharmaceuticals.
But the technique is not restricted to just small crystals.
"The method we developed can handle small protein crystals, but it can also be used for any size protein crystals, any time you need to combine data from more than one sample, "Liu disse.