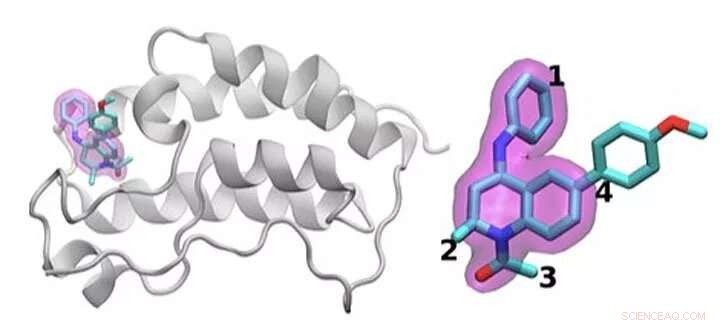
Um esquema da proteína BRD4 ligada a um dos 16 medicamentos com base no mesmo arcabouço de tetraidroquinolina (destacado em magenta). As regiões que são quimicamente modificadas entre as drogas investigadas neste estudo são rotuladas de 1 a 4. Normalmente, apenas uma pequena mudança é feita na estrutura química de um medicamento para o outro. Essa abordagem conservadora permite que os pesquisadores explorem por que um medicamento é eficaz e outro não. Crédito:Laboratório Nacional de Brookhaven
Identificar o tratamento medicamentoso ideal é como atingir um alvo em movimento. Para parar a doença, drogas de pequenas moléculas se ligam fortemente a uma proteína importante, bloqueando seus efeitos no corpo. Mesmo os medicamentos aprovados geralmente não funcionam em todos os pacientes. E com o tempo, agentes infecciosos ou células cancerosas podem sofrer mutação, tornando inútil uma droga outrora eficaz.
Um problema físico fundamental está por trás de todas essas questões:otimizar a interação entre a molécula do medicamento e sua proteína alvo. As variações nas moléculas candidatas a drogas, a faixa de mutação nas proteínas e a complexidade geral dessas interações físicas tornam esse trabalho difícil.
Shantenu Jha, do Laboratório Nacional Brookhaven e da Universidade Rutgers, do Departamento de Energia (DOE), lidera uma equipe que tenta otimizar os métodos computacionais para que os supercomputadores possam assumir parte dessa imensa carga de trabalho. Eles descobriram uma nova estratégia para lidar com uma parte:diferenciar como os candidatos a medicamentos interagem e se ligam a uma proteína-alvo.
Por seu trabalho, Jha e seus colegas ganharam o prêmio IEEE International Scalable Computing Challenge (SCALE) do ano passado, que reconhece soluções de computação escalonáveis para problemas de ciência e engenharia do mundo real.
Para projetar um novo medicamento, uma empresa farmacêutica pode começar com uma biblioteca de milhões de moléculas candidatas que elas reduzem aos milhares que mostram alguma ligação inicial a uma proteína-alvo. Refinar essas opções em uma droga útil que pode ser testada em humanos pode envolver experimentos extensivos para adicionar ou subtrair grupos de átomos em locais-chave na molécula e testar como cada uma dessas mudanças altera como a pequena molécula e a proteína interagem.
Simulações podem ajudar neste processo. Maior, supercomputadores mais rápidos e algoritmos cada vez mais sofisticados podem incorporar física realista e calcular as energias de ligação entre várias moléculas pequenas e proteínas. Esses métodos podem consumir recursos computacionais significativos, Contudo, para obter a precisão necessária. Simulações úteis para a indústria também devem fornecer respostas rápidas. Por causa do cabo de guerra entre precisão e velocidade, pesquisadores estão constantemente inovando, desenvolver algoritmos mais eficientes e melhorar o desempenho, Jha diz.
Esse problema também requer o gerenciamento de recursos computacionais de maneira diferente do que para muitos outros problemas de grande escala. Em vez de projetar uma única simulação que pode ser dimensionada para usar um supercomputador inteiro, pesquisadores executam simultaneamente muitos modelos menores que moldam uns aos outros e a trajetória de cálculos futuros, uma estratégia conhecida como computação baseada em conjunto, ou fluxos de trabalho complexos.
"Pense nisso como tentar explorar uma grande paisagem aberta para tentar encontrar onde você pode ser capaz de obter o melhor candidato a medicamento, "Jha diz. No passado, pesquisadores pediram aos computadores que navegassem neste cenário fazendo escolhas estatísticas aleatórias. Em um ponto de decisão, metade dos cálculos pode seguir um caminho, a outra metade outra.
Jha e sua equipe procuram maneiras de ajudar essas simulações a aprender com a paisagem. Ingerir e compartilhar dados em tempo real não é fácil, Jha diz, "e isso é o que exigia que algumas das inovações tecnológicas fossem feitas em escala." Ele e sua equipe baseada em Rutgers estão colaborando com o grupo de Peter Coveney na University College London neste trabalho.
Para testar essa ideia, eles usaram algoritmos que prevêem a afinidade de ligação e introduziram versões simplificadas em uma estrutura HTBAC, para calculadora de afinidade de ligação de alto rendimento. Uma dessas calculadoras, conhecido como ESMACS, ajuda a eliminar moléculas que se ligam mal a uma proteína-alvo. O outro, LAÇOS, é mais preciso, mas mais limitado em escopo e requer 2,5 vezes mais recursos computacionais. Apesar disso, pode ajudar os pesquisadores a otimizar uma interação promissora entre um medicamento e uma proteína. A estrutura HTBAC os ajuda a implementar esses algoritmos de forma eficiente, salvando o algoritmo mais intensivo para situações em que é necessário.
A equipe demonstrou a ideia examinando 16 candidatos a medicamentos de uma biblioteca de moléculas da GlaxoSmithKline (GSK) com seu alvo, BRD4-BD1 - uma proteína importante no câncer de mama e em doenças inflamatórias. As drogas candidatas tinham a mesma estrutura central, mas diferiam em quatro áreas distintas ao redor das bordas da molécula.
Neste estudo inicial, a equipe executou milhares de processos simultaneamente em 32, 000 núcleos em águas azuis, um supercomputador da National Science Foundation (NSF) na Universidade de Illinois em Urbana-Champaign. Eles fizeram cálculos semelhantes em Titan, o supercomputador Cray XK7 no Oak Ridge Leadership Computing Facility, uma facilidade de usuário do DOE Office of Science. A equipe distinguiu com sucesso entre a ligação desses 16 candidatos a drogas, a maior simulação até hoje. "Não atingimos apenas uma escala sem precedentes, "Jha diz." Nossa abordagem mostra a capacidade de diferenciar. "
Eles ganharam o prêmio SCALE por esta prova de conceito inicial. O desafio agora, Jha diz, está certificando-se de que não funcione apenas para BRD4, mas também para outras combinações de moléculas de drogas e alvos de proteína.
Se os pesquisadores puderem continuar a expandir sua abordagem, essas técnicas podem, eventualmente, ajudar a acelerar a descoberta de medicamentos e possibilitar a medicina personalizada. Mas para examinar problemas mais realistas, eles precisarão de mais poder computacional. "Estamos no meio desta tensão entre um espaço químico muito grande que nós, em princípio, precisa explorar, e, infelizmente, recursos de computador limitados. "Jha diz.
Mesmo quando a supercomputação se expande em direção à exascale, os cientistas da computação podem mais do que preencher a lacuna adicionando física mais realista a seus modelos. Para um futuro próximo, os pesquisadores precisarão ser engenhosos para aumentar esses cálculos. A necessidade é a mãe da inovação, Jha diz, justamente porque a ciência molecular não terá a quantidade ideal de recursos computacionais para fazer simulações.
Mas a computação exascale pode ajudar a aproximá-los de seus objetivos. Além de trabalhar com a University College London e GSK, Jha e seus colegas estão colaborando com Rick Stevens, do Argonne National Laboratory e com a equipe do CANcer Distributed Learning Environment (CANDLE). Este projeto de co-design dentro do Projeto de Computação Exascale do DOE está construindo redes neurais profundas e técnicas gerais de aprendizado de máquina para estudar o câncer. Os algoritmos e software dentro do HTBAC podem complementar o foco do CANDLE nessas abordagens.
Esta colaboração mais ampla entre o grupo de Jha, a equipe CANDLE e o laboratório de John Chodera no Memorial Sloan-Kettering Cancer Center lideraram o projeto de previsão de resistência integrada e escalonável (INSPIRE). Esta equipe já fez simulações no supercomputador Summit do DOE no Oak Ridge National Laboratory. Em breve, ela continuará esse trabalho no Frontera - a máquina de liderança da NSF na Universidade do Texas no Texas Advanced Computing Center de Austin.
"Estamos ansiosos por um maior progresso e maiores aprimoramentos metodológicos, "Jha diz." Gostaríamos de ver como essas abordagens bastante complementares podem funcionar de forma integrada em direção a essa grande visão. "