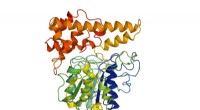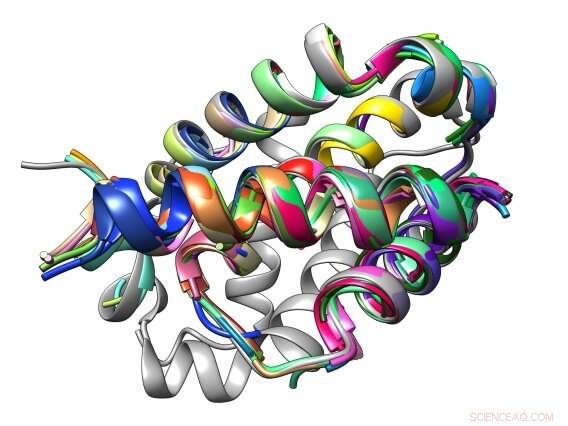
A interface de ligação entre um peptídeo e sua proteína alvo Bcl-2 é composta de motivos estruturais comuns conhecidos como TERMs. Crédito:Sebastian Swanson e Avi Singer
Uma maneira de investigar sistemas biológicos complexos é bloquear a interação de seus componentes e ver o que acontece. Este método permite que os pesquisadores entendam melhor os processos e funções celulares, aumentando os experimentos diários de laboratório, ensaios diagnósticos, e intervenções terapêuticas. Como resultado, reagentes que impedem as interações entre as proteínas estão em alta demanda. Mas antes que os cientistas possam gerar rapidamente suas próprias moléculas personalizadas capazes de fazer isso, eles devem primeiro analisar a relação complicada entre sequência e estrutura.
Moléculas pequenas podem entrar nas células facilmente, mas a interface onde duas proteínas se ligam uma à outra costuma ser muito grande ou não tem as minúsculas cavidades necessárias para que essas moléculas tenham como alvo. Anticorpos e nanocorpos se ligam a trechos mais longos de proteína, o que os torna mais adequados para impedir as interações proteína-proteína, mas seu grande tamanho e estrutura complexa tornam-nos difíceis de aplicar e instáveis no citoplasma. Por contraste, trechos curtos de aminoácidos, conhecidos como peptídeos, são grandes o suficiente para ligar longos trechos de proteína, embora ainda sejam pequenos o suficiente para entrar nas células.
O laboratório Keating no Departamento de Biologia do MIT está trabalhando duro para desenvolver maneiras de criar peptídeos rapidamente que podem interromper as interações proteína-proteína envolvendo proteínas Bcl-2, que promovem o crescimento do câncer. Sua abordagem mais recente utiliza um programa de computador chamado dTERMen, desenvolvido por ex-alunos do laboratório Keating, Gevorg Grigoryan Ph.D. '07, atualmente é professor associado de ciência da computação e professor associado adjunto de ciências biológicas e química no Dartmouth College. Os pesquisadores simplesmente alimentam o programa com as estruturas desejadas, e cospe sequências de aminoácidos para peptídeos capazes de interromper interações proteína-proteína específicas.
"É uma abordagem tão simples de usar, "diz Keating, professor de biologia do MIT e autor sênior do estudo. "Em teoria, você poderia colocar em qualquer estrutura e resolver para uma sequência. Em nosso estudo, o programa surgiu com novas combinações de sequência que não se parecem com nada encontrado na natureza - ele deduziu uma maneira completamente única de resolver o problema. É emocionante descobrir novos territórios do universo da sequência. "
O ex-pós-doutorado Vincent Frappier e Justin Jenson Ph.D. '18 são co-primeiros autores do estudo, que aparece na última edição de Estrutura .
Mesmo problema, abordagem diferente
Jenson, para a parte dele, enfrentou o desafio de projetar peptídeos que se ligam às proteínas Bcl-2 usando três abordagens distintas. O método baseado em dTERMen, ele diz, é de longe o mais eficiente e geral que ele já experimentou.
Abordagens padrão para descobrir inibidores de peptídeos geralmente envolvem modelar moléculas inteiras até a física e a química por trás dos átomos individuais e suas forças. Outros métodos requerem telas demoradas para os melhores candidatos de ligação. Em ambos os casos, o processo é árduo e a taxa de sucesso é baixa.
dTERMen, por contraste, não necessita de física nem de triagem experimental, e aproveita unidades comuns de estruturas de proteínas conhecidas, como hélices alfa e fitas beta - chamadas de motivos estruturais terciários ou "TERMs" - que são compiladas em coleções como o Protein Data Bank. dTERMen extrai esses elementos estruturais do banco de dados e os usa para calcular quais sequências de aminoácidos podem adotar uma estrutura capaz de se ligar e interromper interações proteína-proteína específicas. Leva um único dia para construir o modelo, e meros segundos para avaliar mil sequências ou projetar um novo peptídeo.
"dTERMen nos permite encontrar sequências que provavelmente têm as propriedades de ligação que procuramos, em um robusto, eficiente, e de maneira geral com uma alta taxa de sucesso, "Jenson diz." Abordagens anteriores levaram anos. Mas usando dTERMen, passamos de estruturas para projetos validados em questão de semanas. "
Dos 17 peptídeos que eles construíram usando as sequências projetadas, 15 ligado com afinidade de tipo nativo, interromper as interações proteína-proteína Bcl-2 que são notoriamente difíceis de atingir. Em alguns casos, seus designs eram surpreendentemente seletivos e vinculados a um único membro da família Bcl-2 em detrimento dos outros. As sequências projetadas desviaram-se de sequências conhecidas encontradas na natureza, o que aumenta muito o número de peptídeos possíveis.
“Este método permite um certo nível de flexibilidade, "Frappier diz." DTERMen é mais robusto para mudanças estruturais, o que nos permite explorar novos tipos de estruturas e diversificar nosso portfólio de candidatos potenciais de ligação. "
Sondando o universo da sequência
Dados os benefícios terapêuticos de inibir a função de Bcl-2 e retardar o crescimento do tumor, o laboratório Keating já começou a estender seus cálculos de projeto para outros membros da família Bcl-2. Eles pretendem eventualmente desenvolver novas proteínas que adotem estruturas nunca vistas antes.
"Já vimos exemplos suficientes de várias estruturas de proteínas locais que modelos computacionais de relações sequência-estrutura podem ser inferidos diretamente de dados estruturais, em vez de ter que ser redescoberto a cada vez a partir de princípios de interação atomística, "diz Grigoryan, Criador do dTERMen. "É imensamente empolgante que essa inferência baseada em estrutura funcione e seja precisa o suficiente para permitir um projeto de proteína robusto. Ele fornece uma ferramenta fundamentalmente diferente para ajudar a resolver os principais problemas da biologia estrutural - do projeto da proteína à previsão da estrutura."
Frappier espera um dia ser capaz de rastrear todo o proteoma humano computacionalmente, usando métodos como dTERMen para gerar peptídeos de ligação candidatos. Jenson sugere que o uso de dTERMen em combinação com abordagens mais tradicionais para redesenho de sequência pode ampliar uma ferramenta já poderosa, capacitando os pesquisadores a produzir esses peptídeos direcionados. Idealmente, ele diz, um dia, desenvolver peptídeos que se ligam e inibem sua proteína favorita pode ser tão fácil quanto executar um programa de computador, ou tão rotineiro quanto projetar um primer de DNA.
De acordo com Keating, embora esse tempo ainda esteja no futuro, "nosso estudo é o primeiro passo para demonstrar essa capacidade em um problema de alcance modesto."
Esta história foi republicada por cortesia do MIT News (web.mit.edu/newsoffice/), um site popular que cobre notícias sobre pesquisas do MIT, inovação e ensino.