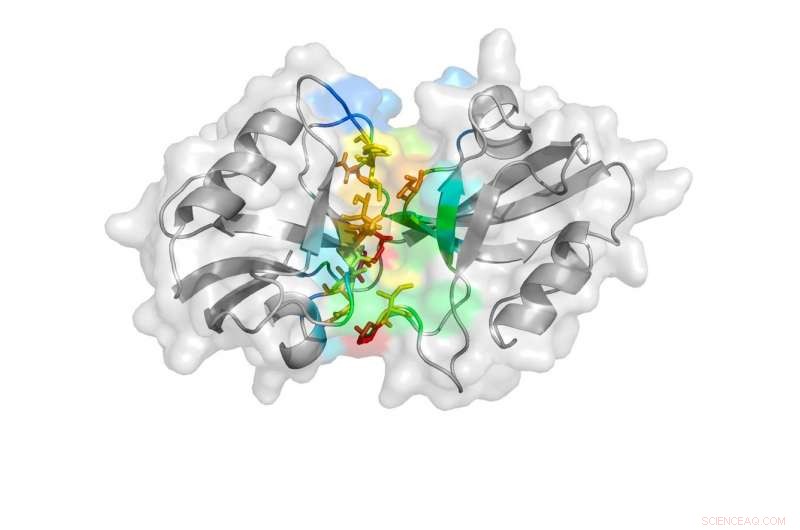
Uma representação de cartoon revelada pelo estudo mostra o estado de tipo fechado dos domínios PDZ. Crédito: Nature Communications , doi:10.1038 / s41467-018-06133-0
Em uma extensão de pesquisa publicada há um mês em Métodos da Natureza , uma nova abordagem híbrida realizada por pesquisadores do departamento de física e astronomia da Clemson University e da Stony Brook University revelou uma estrutura 3-D de um fragmento de proteína que poderia servir como um alvo de droga no tratamento de pacientes com derrame.
A proteína chamada "proteína de densidade pós-sináptica de 95 kDa (PSD-95)" está posicionada nos neurônios do cérebro que estão recebendo mensagens químicas - neurotransmissores - de neurônios adjacentes. Ao recrutar receptores e outras proteínas auxiliares, O PSD-95 trabalha para manter a integridade das conexões neurais ao longo do tempo, facilitando assim a comunicação neural, aprendizagem e memória.
PSD-95 consiste em cinco partes, ou domínios, que cada um desempenha um papel diferente na função geral da proteína. Dois desses domínios, chamado PDZ-1 e PDZ-2, mostraram influenciar os sintomas associados ao AVC isquêmico, como paralisia ou dificuldade de fala.
"Uma das ideias postuladas na literatura é criar uma droga multivalente que atinja ambos os domínios PDZ porque eles são muito semelhantes em natureza. Se você pode bloquear os domínios PDZ de ligação a proteínas ou enzimas específicas, você pode reduzir os efeitos debilitantes de um derrame, "disse Hugo Sanabria, autor principal do estudo.
O desafio, Contudo, é que é quase impossível projetar um inibidor de drogas sem primeiro saber a estrutura exata dos domínios PDZ do PSD-95. Seria como dirigir pelo país sem ter um mapa dos Estados Unidos.
"As funções biológicas das biomoléculas são determinadas por suas estruturas, portanto, precisamos de percepções estruturais e dinâmicas detalhadas de PDZ-1 e -2 para ajudar a entender melhor seus papéis funcionais e auxiliar no projeto de novos inibidores, "disse Feng Ding, Colega de Sanabria aqui na Clemson.
Existem várias abordagens para processar a estrutura das biomoléculas. Mas no caso do PSD-95, cada abordagem - espectroscopia de NMR, A cristalografia de raios-X e a transferência de energia de ressonância Förster (FRET) - entregaram um modelo estrutural diferente. O colaborador dos pesquisadores na Stony Brook University, professor associado Mark Bowen no departamento de fisiologia e biofísica, estabeleceu uma parceria com a Sanabria neste projeto depois que ele descobriu um dos modelos estruturais inconsistentes do fragmento PSD-95.
O laboratório de Sanabria tratou dessa discrepância modelando primeiro o fragmento PSD-95 usando FRET, uma abordagem que identifica possíveis configurações de biomoléculas. Sob este método, Sanabria anexou duas moléculas sensíveis à luz, chamados cromóforos, em duas posições diferentes no fragmento PSD-95. Ele então descobriu a distância entre os cromóforos visualizando o fragmento em um microscópio. Isso foi repetido várias vezes em diferentes pontos de fixação.
“Para o aspecto de modelagem, FRET fornece distâncias entre cromóforos, mas isso não é suficiente para preencher todas as restrições geométricas da molécula, então temos que confiar em outra coisa, alguma outra metodologia. É aí que o professor Ding entra em jogo, "Sanabria disse.
Ding lidera um laboratório de biofísica computacional na Clemson University, onde usa um software de computador para avaliar a aparência das biomoléculas, mover e funcionar. Sua abordagem de modelagem utiliza uma simulação de computador conhecida como dinâmica molecular discreta (DMD) que mapeia a paisagem de uma biomolécula, prever as trajetórias das proteínas conforme elas se dobram e interagem com outras moléculas. A simulação subsequente pode ser reproduzida como um filme, ajudando os pesquisadores a visualizar os comportamentos das proteínas ao longo do tempo.
"Se você fizer simulações moleculares tradicionais, normalmente você vai amostrar uma região muito pequena do espaço, particularmente para moléculas maiores, então você não terá uma boa visão geral de como a molécula inteira ficará, mesmo em condições fisiológicas, "Sanabria disse." A dinâmica molecular discreta é uma maneira muito mais rápida e menos dispendiosa do ponto de vista computacional de amostrar com precisão e rapidez o espaço conformacional das proteínas. "
Para fazer isso, Sanabria primeiro obteve um conjunto de distâncias medindo PSD-95 com FRET. Nesse experimento, Sanabria tinha 10 amostras do fragmento PSD-95, cada uma reproduzindo distâncias diferentes e três formas comuns - ou conformações - de PSD-95 foram observadas. Ainda, sem uma simulação DMD, não havia como os pesquisadores saberem qual distância correspondia a qual conformação do fragmento. Assim, eles inserem cada distância possível em relação a cada forma possível e deixam a simulação fazer o resto.
"Assim que fizemos a primeira simulação, vimos que havia três estados principais que PDZ-1 e -2 estavam tomando. Um mostrou um contato muito próximo entre os dois, um mostrou um conjunto de contato intermediário e outro não teve nenhum contato, "Ding disse.
Os pesquisadores então executaram uma simulação DMD novamente sem considerar as distâncias FRET para confirmar que os três estados observados existem na natureza e não são simplesmente um golpe de sorte imposto pelas distâncias FRET. Eles investigaram ainda mais as estruturas, observando a maneira como os aminoácidos individuais, que constituem os domínios PDZ, vínculo um com o outro. A partir dessas análises, Ding, Bowen e Sanabria foram capazes de confirmar que os domínios PDZ assumem dois dos três estados observados na simulação DMD - aquele com algum contato e aquele sem nenhum contato.
"Agora, temos dois alvos potenciais para a engenharia de novos medicamentos que serão mais eficientes do que os que estão disponíveis atualmente, "Sanabria disse." As perspectivas para pacientes com derrame são promissoras.
Sem dinâmica molecular discreta, que pode capturar mudanças conformacionais que ocorrem na escala de tempo de microssegundos, esses dois estados teriam sido perdidos como em estudos anteriores.
"A maioria das pessoas que fazem modelagem estrutural guiada por FRET estão trabalhando com uma molécula rígida, como DNA. Se você tem uma molécula rígida, é fácil de modelar - você tem apenas um único estado para capturar. Você pode atribuir as distâncias FRET e não há realmente nenhum problema, "Sanabria disse." Neste caso, ultrapassamos essa abordagem de várias maneiras. "
Em estudos futuros, a equipe está procurando analisar o potencial do fragmento PSD-95 de se auto-inibir com base na própria estrutura do fragmento.