Os pesquisadores da Rice University e do Baylor College of Medicine usaram simulações de computador para estudar o processo pelo qual a hemaglutinina ajuda os vírus a invadir e infectar as células. Os pesquisadores acreditam que o domínio do tronco da proteína se desdobra e se desdobra em uma configuração diferente quando acionado, mas faz uma pausa para liberar um peptídeo de fusão oculto que liga o vírus à célula-alvo. Clique na imagem para ver uma versão maior. Crédito:Xingcheng Lin
Há um obstáculo no balanço de uma proteína que transmite o vírus da gripe. Os pesquisadores da Rice University e do Baylor College of Medicine acreditam que esse mecanismo pode ser um alvo útil para impedir que o vírus infecte as células.
Em um artigo no Proceedings of the National Academy of Sciences, a equipe Rice-Baylor liderada pelo biofísico José Onuchic e os bioquímicos Jianpeng Ma e Qinghua Wang investiga mais profundamente um complexo de glicoproteínas que começou a definir em um artigo de 2014.
Essa proteína, hemaglutinina, fica na superfície dos vírus da gripe e os ajuda a se anexar e transportar através das membranas protetoras das células-alvo.
O artigo começa a definir o mecanismo que permite à proteína se desdobrar e redobrar em um piscar de olhos, mudando sua forma para expor um peptídeo que anexa o vírus a uma célula e inicia a infecção. Os pesquisadores acreditam que as drogas terapêuticas podem usar esse mecanismo para desligar o vírus.
"Esta proteína começa em um estado dobrado e passa por uma transformação global, redobrando em um estado completamente diferente, "disse Onuchic, co-diretor do Centro de Física Teórica Biológica de Rice (CTBP). "Mas há uma pequena parte no centro que a evolução conservou."
Esse único resíduo de aminoácido conservado é o obstáculo que faz a proteína parar no processo de redobramento. Ele permite que um peptídeo de fusão enterrado no interior se ligue à célula-alvo e comece a infectá-la. Sem pausa, a redobragem seria muito rápida para que a ligação ocorresse.
O autor principal e pesquisador de pós-doutorado de Rice, Xingcheng Lin, modelou essa parte da proteína, o B-loop do domínio HA2. HA2 fica abaixo de outro domínio, um limite conhecido como HA1 que sofre mutação para escapar de defesas passadas. Lin explicou que HA1 é um alvo comum para medicamentos contra gripe porque o domínio cap exposto é mais acessível do que o domínio HA2 protegido.
O problema é que o HA1 sofre mutação constante para resistir às drogas, ele disse. Isso influencia a eficácia das vacinas contra a gripe todos os anos. Lin e Onuchic disseram que HA2 apresenta um alvo melhor para drogas porque o mecanismo é altamente conservado pela evolução.
“Se um medicamento tem como alvo HA2, o domínio não pode escapar fazendo mutações porque as próprias mutações o tornariam não funcional, "Lin disse." Esse tipo de droga pode se tornar uma vacina universal.
HA2 é uma estrutura trimérica que, quando acionado por condições ácidas no ambiente perto de uma célula-alvo, se transforma de um loop aleatório em uma bobina enrolada. Mesmo com a pausa, ele se desdobra e se desdobra em uma fração de segundo, muito rápido para os microscópios verem. Mas uma simulação de computador do processo pode ser retardada.
Isso passa a ser uma especialidade do CTBP, que usa programas que analisam o panorama de energia das proteínas para prever como elas se dobrarão. Onuchic e seus colegas são pioneiros na teoria de que o dobramento de proteínas segue uma ordem, processo "afunilado" que depende da energia intrínseca de cada átomo da cadeia, cada um deles busca constantemente seu estado de energia mais baixo. Se todas as "contas" atômicas puderem ser identificadas, é possível simular o complexo processo de dobramento.
Os pesquisadores do Rice costumam usar modelos de proteínas de granulação grossa, um subconjunto de átomos que representam o todo, para prever como eles irão desistir. O novo estudo foi muito mais ambicioso e estabelecido para prever o desdobramento e redobramento complexo, usando não apenas cada átomo da cadeia, mas também cada átomo em seu ambiente líquido, Onuchic disse.
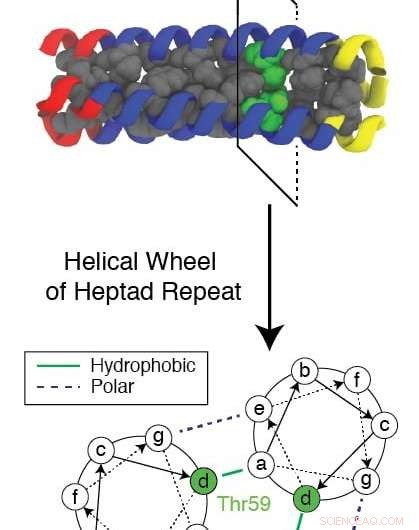
Um resíduo conservado evolucionário conhecido como Thr59 interrompe o padrão de repetição formado por uma proteína trimérica à medida que ela se dobra ao mesmo tempo em que ajuda um vírus da gripe a infectar uma célula. Pesquisadores da Rice University e do Baylor College of Medicine usaram uma complexa simulação de computador para estudar o mecanismo e procurar novos alvos para medicamentos para parar a gripe. Crédito:Xingcheng Lin
Lin modelou 40 microssegundos (milionésimos de segundo) da transição do domínio HA2 que representa todo o processo, que leva 1,4 milissegundos (milésimos de segundo) para ser concluído. Mesmo esse processo encurtado levou dois anos do tempo do computador para entregar resultados, ele disse.
"O domínio simulado é de cerca de 3, 000 átomos, mas quando o ambiente, incluindo água, é contabilizado, a simulação total incorpora cerca de 100, 000 átomos, "Onuchic disse." Ainda é uma simulação enorme que exigia técnicas de ponta. "
Teorias anteriores baseadas em imagens cristalográficas das proteínas antes e depois propunham a ideia de um domínio com mola que parecia se ligar à célula-alvo após a remoção da capa. Onuchic disse que o modelo completo do HA2 suporta um mecanismo diferente.
"Descobrimos que há um monte de energia que torna o estado final do HA2 muito mais estável do que o estado inicial, "disse ele." Mas com o mecanismo de mola, a maior parte da energia já seria desperdiçada no momento em que ela forma a bobina enrolada e se liga à célula e às membranas virais. Não deixaria nenhuma energia para juntar as membranas.
"É por isso que decidimos fazer um cálculo completo do sistema - todos os átomos da proteína e toda a água, "Onuchic disse." Foi um esforço gigantesco.
O resíduo hidrofílico conservado (atração de água), conhecido como Thr59, é de particular interesse para os pesquisadores não apenas pela maneira como interrompe o dobramento e permite que o vírus ataque, mas também porque tem um irmão gêmeo.
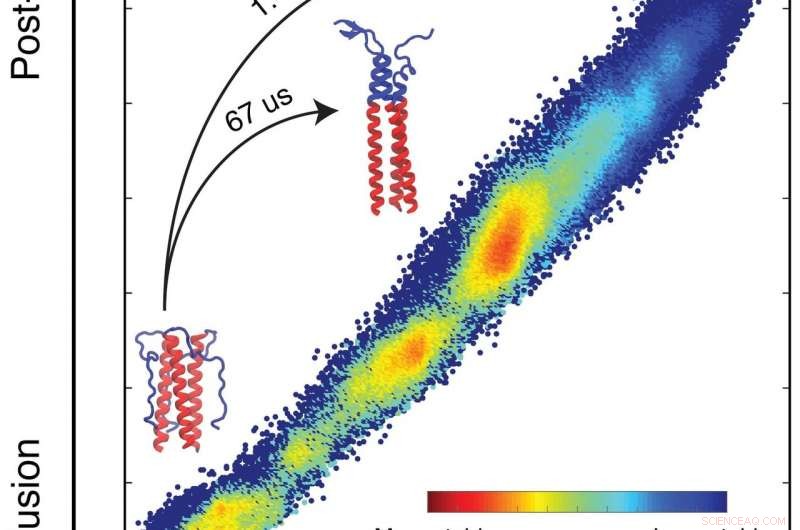
Uma simulação feita por biofísicos da Rice University detalhou o perfil de energia livre que dita como uma proteína que ajuda o vírus da gripe a infectar as células cumpre sua missão. As simulações prevêem como uma proteína se dobrará com base nas energias intrínsecas de cada átomo no sistema. As proteínas formam loops e bobinas à medida que buscam seu nível mais baixo, estados de energia mais estáveis (azul). No domínio que os pesquisadores estudaram, eles encontraram um obstáculo que retarda o processo de dobramento que permite a ligação à célula-alvo e também apresenta uma oportunidade para novas vacinas atacarem a gripe. Clique na imagem para ver uma versão maior. Crédito:Xingcheng Lin
"Na árvore evolutiva completa, esses vírus se dividem em dois grupos, e a diferença parece ser este resíduo, "Onuchic disse." Eles se dividem em 1, 500 anos atrás e de alguma forma, após esta separação, eles estão totalmente conservados. Eles não foram capazes de mudar esse resíduo, não importa o que aconteça, e acreditamos que isso torna esse resíduo importante. "
A pesquisa atual se concentrou no grupo que incorpora Thr59 e causa a cepa H3N2 responsável pela gripe de Hong Kong, Lin disse. O outro resíduo, Met59, aparece na cepa H1N1 que causou a gripe espanhola.
"Ainda temos um longo caminho a percorrer para entender toda a proteína, "disse ele." Aqui, nós apenas estudamos um domínio de uma proteína, e há vários outros que são muito importantes para sua função. "
"Mas o que Xingcheng já fez é um tour de force computacional, "Onuchic acrescentou." Ele mostrou como este resíduo particular quebra a simetria helicoidal do domínio e o torna instável o suficiente para dar ao peptídeo tempo para agarrar as membranas. "