Crédito:Universidade de Michigan
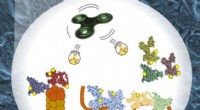
As proteínas são as abelhas operárias da célula, principalmente agrupando-se para formar macromoleculares, complexos multicomponentes para realizar tarefas celulares complexas.
Tentar caracterizar esses complexos de proteínas e todas as suas funções dentro dos organismos é uma disciplina chamada "proteômica". Historicamente, os cientistas estudaram a forma e a função das proteínas, dissolvendo-as com enzimas e sequenciando os pequenos pedaços quebrados resultantes, chamados de peptídeos. Mas estudar proteínas dessa forma causa a perda de muitas informações essenciais, diz Brandon Ruotolo, professor associado de química da Universidade de Michigan.
Ruotolo e sua equipe, incluindo pesquisadores da UCLA, University of Leeds e University of Antwerp, desenvolveram uma nova maneira de estudar os complexos de proteínas que não envolvem a destruição dos conjuntos intactos no processo. Seu método foi publicado na revista Química Analítica .
"As proteínas são os principais impulsionadores de praticamente todos os processos celulares críticos - tudo, desde a divisão celular até a morte celular, “Disse Ruotolo.“ Eles também dominam os alvos das drogas por causa de sua importância central no contexto da vida como a conhecemos. Entender como essas proteínas funcionam, composicionalmente, em um nível muito básico, é muito importante para entender como as doenças funcionam. "
Tanto a abordagem tradicional quanto o método de Ruotolo usam um dispositivo chamado espectrômetro de massa, que mede o peso, ou massa, de moléculas ionizadas puxando as moléculas para o vácuo. Na abordagem tradicional, depois que os cientistas quebraram os complexos de proteínas em peptídeos, eles usam uma tecnologia chamada ionização por eletropulverização para dar a esses peptídeos uma carga elétrica. O espectrômetro de massa então mede a massa desses peptídeos carregados, e os decompõe ainda mais usando um gás de fundo.
Mas usar enzimas para digerir proteínas torna difícil entender o papel de entidades químicas menores que se acumulam nas proteínas, chamadas modificações pós-traducionais. Cada vez que sua célula expressa uma proteína, também pode produzir centenas desses estados individuais de modificação pós-tradução, chamados coletivamente de proteoformas, Ruotolo diz.
É o arranjo dessas proteoformas dentro dos complexos de proteínas que freqüentemente determina sua função. Em abordagens tradicionais para estudar complexos de proteínas, essas proteoformas são perdidas.
"Os principais motores e agitadores na célula não são proteínas individuais correndo por aí fazendo trabalhos - na verdade, são dezenas de proteínas que se juntam para formar complexos supermoleculares que realizam trabalhos muito complicados na célula, "Ruotolo disse." Agora, a verdadeira tarefa é entender como funcionam essas grandes máquinas. "
A equipe de Ruotolo usa ionização por eletrospray para ionizar complexos protéicos intactos. Como os peptídeos normalmente analisados em estudos de proteômica, esses complexos multiproteicos podem ser sequenciados por um espectrômetro de massa, mas geralmente não é possível sequenciar nada mais do que uma pequena fração da estrutura da montagem. A equipe de Ruotolo desenvolveu uma estratégia de modificação química que melhora significativamente a capacidade de sequenciar grandes, complexos multiproteicos diretamente por espectrometria de massa.
"Isso é importante porque no espectrômetro de massa, você tem conectividade entre a proteoforma inicial e os íons de sequência, "Ruotolo disse." Na digestão enzimática, que a conectividade está interrompida. "
Em seu estudo, A equipe de Ruotolo desenvolveu seu método usando três complexos de proteínas diferentes. Eles esperam adaptar seu método para poder estudar complexos de proteínas maiores, muitas vezes o tamanho daqueles em seu estudo.
"Acho que estamos aprendendo todos os dias que mesmo uma classe de câncer, como leucemia, é composto de muitas doenças diferentes, "Ruotolo disse." Os tipos de medições que estamos desenvolvendo só irão aumentar nossa capacidade de observar essa granularidade e fornecer informações que podem, esperançosamente, informar a capacidade de descobrir novas terapias. "
O grupo também publicou um artigo em Química Analítica detalhando software, que foi desenvolvido em colaboração com pesquisadores do Departamento de Medicina Computacional e Bioinformática da U-M. O software é capaz de capturar rapidamente informações de sequência de complexos de proteínas.