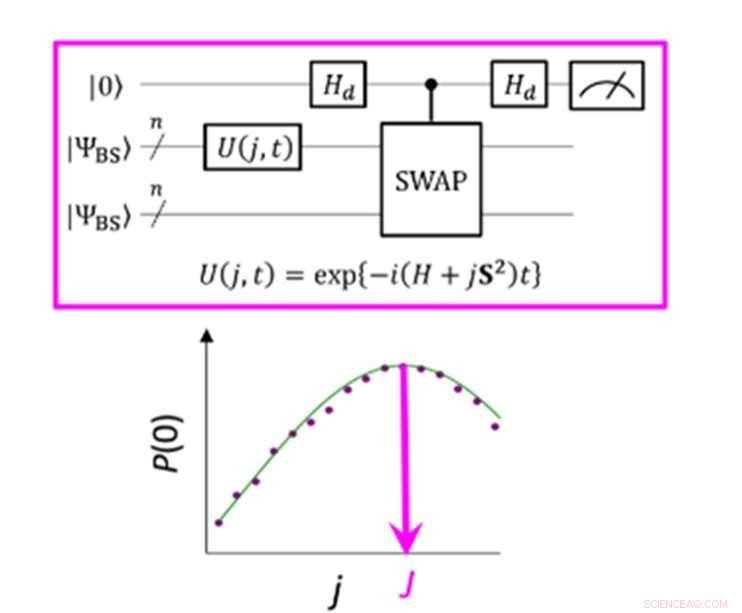
 p Um circuito quântico que permite a probabilidade máxima de P (0) na medição do parâmetro J. Crédito:K. Sugisaki, K. Sato e T. Takui
p Um circuito quântico que permite a probabilidade máxima de P (0) na medição do parâmetro J. Crédito:K. Sugisaki, K. Sato e T. Takui
p Pesquisadores da Universidade da Cidade de Osaka usam estados de superposição quântica e inferência Bayesiana para criar um algoritmo quântico, facilmente executável em computadores quânticos, que calcula com precisão e diretamente as diferenças de energia entre o solo eletrônico e os estados de spin excitados de sistemas moleculares em tempo polinomial. p Compreender como o mundo natural funciona nos permite imitá-lo para o benefício da humanidade. Pense no quanto dependemos de baterias. No centro está a compreensão das estruturas moleculares e do comportamento dos elétrons dentro delas. Calcular as diferenças de energia entre o solo eletrônico de uma molécula e os estados de spin excitados nos ajuda a entender como usar melhor essa molécula em uma variedade de produtos químicos, aplicações biomédicas e industriais. Fizemos muito progresso em moléculas com sistemas de casca fechada, em que os elétrons estão emparelhados e estáveis. Sistemas de shell aberto, por outro lado, são menos estáveis e seu comportamento eletrônico subjacente é complexo, e, portanto, mais difícil de entender. Eles têm elétrons desemparelhados em seu estado fundamental, que fazem com que sua energia varie devido à natureza intrínseca dos spins do elétron, e torna as medições difíceis, especialmente à medida que as moléculas aumentam de tamanho e complexidade. Embora essas moléculas sejam abundantes na natureza, faltam algoritmos que possam lidar com essa complexidade. Um obstáculo tem sido lidar com o que é chamado de explosão exponencial do tempo computacional. Usar um computador convencional para calcular como os spins desemparelhados influenciam a energia de uma molécula de casca aberta levaria centenas de milhões de anos, tempo que os humanos não têm.
p Computadores quânticos estão em desenvolvimento para ajudar a reduzir isso ao que é chamado de "tempo polinomial". Contudo, o processo que os cientistas têm usado para calcular as diferenças de energia das moléculas de casca aberta tem sido essencialmente o mesmo para os computadores convencionais e quânticos. Isso dificulta o uso prático da computação quântica em aplicações químicas e industriais.
p "Abordagens que invocam algoritmos quânticos verdadeiros nos ajudam a tratar sistemas de shell aberto com muito mais eficiência do que utilizando computadores clássicos, "estadual Kenji Sugisaki e Takeji Takui da Universidade da Cidade de Osaka. Com seus colegas, eles desenvolveram um algoritmo quântico executável em computadores quânticos, que pode, pela primeira vez, calcular com precisão as diferenças de energia entre o solo eletrônico e os estados de spin excitados de sistemas moleculares de casca aberta. Suas descobertas foram publicadas no jornal
Ciência Química em 24 de dezembro de 2020.
p A diferença de energia entre os estados de spin molecular é caracterizada pelo valor do parâmetro de interação de troca J. Os algoritmos quânticos convencionais foram capazes de calcular com precisão as energias para moléculas de casca fechada ", mas não foram capazes de lidar com sistemas com uma forte multi-configuração personagem, "afirma o grupo. Até agora, os cientistas presumiram que, para obter o parâmetro J, deve-se primeiro calcular a energia total de cada estado de spin. Em moléculas de casca aberta, isso é difícil porque a energia total de cada estado de spin varia muito à medida que a atividade e o tamanho da molécula mudam. Contudo, "a diferença de energia em si não depende muito do tamanho do sistema, "observa a equipe de pesquisa. Isso os levou a criar um algoritmo com cálculos que focavam na diferença de spin, não os estados de spin individuais. A criação de tal algoritmo exigiu que eles abandonassem as suposições desenvolvidas a partir de anos de uso de computadores convencionais e se concentrassem nas características únicas da computação quântica - a saber, "estados de superposição quântica".
p "Superposição" permite que os algoritmos representem duas variáveis ao mesmo tempo, o que então permite que os cientistas se concentrem na relação entre essas variáveis sem qualquer necessidade de determinar seus estados individuais primeiro. A equipe de pesquisa usou algo chamado de função de onda de simetria quebrada como uma superposição de funções de onda com diferentes estados de spin e reescreveu na equação hamiltoniana para o parâmetro J. Ao executar este novo circuito quântico, a equipe foi capaz de se concentrar nos desvios de sua meta e aplicando a inferência Bayesiana, uma técnica de aprendizado de máquina, eles trouxeram esses desvios para determinar o parâmetro de interação de troca J. "Simulações numéricas baseadas neste método foram realizadas para a dissociação covalente de hidrogênio molecular (H
2 ), a dissociação da ligação tripla do nitrogênio molecular (N
2 ), e os estados básicos de C, O, Átomos de Si e NH, OH
+
, CH
2 , NF e O
2 moléculas com um erro de menos de 1 kcal / mol, "acrescenta a equipe de pesquisa.
p "Planejamos instalar nossa calculadora de parâmetro de acoplamento Bayesian eXchange com software de funções de onda de simetria quebrada (BxB) em computadores quânticos de curto prazo equipados com dispositivos quânticos ruidosos (sem correção de erro quântico) de escala intermediária (várias centenas de qubits) (dispositivos NISQ ), testando a utilidade para cálculos químicos quânticos de sistemas moleculares consideráveis reais. "