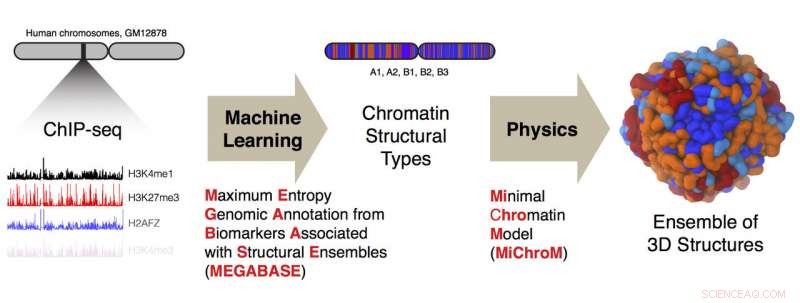
 p Pesquisadores da Rice University e do Baylor College of Medicine desenvolveram um pipeline computacional para converter dados de sequenciamento de ChIP unidimensionais sobre o DNA em estruturas tridimensionais de cromossomos humanos. Crédito:Ryan Cheng / Michele Di Pierro
p Pesquisadores da Rice University e do Baylor College of Medicine desenvolveram um pipeline computacional para converter dados de sequenciamento de ChIP unidimensionais sobre o DNA em estruturas tridimensionais de cromossomos humanos. Crédito:Ryan Cheng / Michele Di Pierro
p O DNA em uma célula humana tem 1,83 metros de comprimento e envolve milhões de proteínas histonas semelhantes a contas para caber no núcleo da célula. Pesquisadores da Rice University e do Baylor College of Medicine mostraram que o exame do estado químico dessas proteínas torna possível prever como um cromossomo de DNA inteiro se dobrará. p Pesquisadores baseados no Centro de Física Teórica Biológica de Rice (CTBP) construíram modelos de computador para analisar marcas epigenéticas, que incluem proteínas ligadas ao DNA, bem como modificações químicas às proteínas histonas. Eles coletaram as informações codificadas nessas marcações para prever como os cromossomos se dobram em três dimensões.
p Suas descobertas aproximam o campo da genética da capacidade de prever a estrutura dobrada de genomas inteiros, que um dia poderia ajudar a identificar doenças genéticas relacionadas ao mal dobramento.
p O trabalho aparece esta semana no
Proceedings of the National Academy of Sciences .
p Empacotado no núcleo, O DNA se dobra em uma forma funcional que difere em vários tipos de células. Como cada célula de um organismo contém o mesmo DNA, marcas epigenéticas ajudam a encontrar a forma certa para o tipo de célula que habita.
p "Algo no topo do código genético diz à célula o que ela deve ser e determina quais partes do cromossomo serão lidas a qualquer momento, "disse o biofísico Peter Wolynes, um co-autor do artigo. "Essas são as chamadas marcas epigenéticas."
p Coletivamente, marcas epigenéticas ajudam a empacotar o genoma nos compartimentos soltos, mas altamente organizados que adota durante a interfase, a "meia-idade" ativa na vida de uma célula. Esses compartimentos aproximam os genes relacionados à transcrição e permitem que eles se comuniquem e funcionem.
p As marcas epigenéticas podem ser reveladas por uma técnica estabelecida chamada sequenciamento ChIP, que mapeia locais de ligação de proteínas ao longo do DNA.
p "Não entendemos exatamente como o genoma é marcado, mas podemos medi-lo por meio de sequenciamento ChIP, que se tornou uma experiência bastante simples, "Wolynes disse." Da mesma forma que podemos ver o código genético (o DNA), também podemos medir essas marcas diretamente em muitas células diferentes. Eles se tornaram a próxima camada de sequência do genoma. "
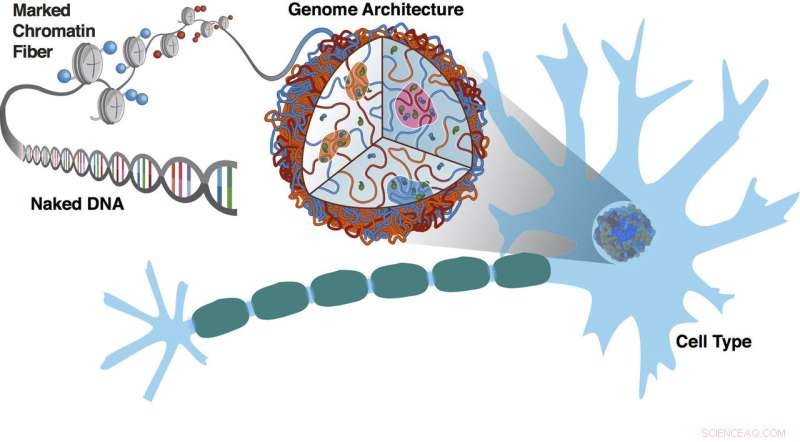 p Experimentos na Rice University e Baylor College of Medicine mostram como segmentos de cromatina com os mesmos padrões de marcação epigenética se localizam juntos em um processo relacionado à separação de fases. O DNA nu é decorado por marcações epigenéticas que codificam o arranjo tridimensional dos cromossomos. A arquitetura do genoma e os padrões de marcação são características do tipo de célula, neste caso, uma célula nervosa com sua bainha de mielina característica. Crédito:Sigrid Knemeyer / Centro de Física Teórica Biológica da Rice University
p Experimentos na Rice University e Baylor College of Medicine mostram como segmentos de cromatina com os mesmos padrões de marcação epigenética se localizam juntos em um processo relacionado à separação de fases. O DNA nu é decorado por marcações epigenéticas que codificam o arranjo tridimensional dos cromossomos. A arquitetura do genoma e os padrões de marcação são características do tipo de célula, neste caso, uma célula nervosa com sua bainha de mielina característica. Crédito:Sigrid Knemeyer / Centro de Física Teórica Biológica da Rice University
p "É outra camada de informação, "disse o coautor e biofísico José Onuchic." Cada um dos DNA de suas células é o mesmo. Contudo, diferentes tipos de células têm diferentes epigenéticas, então seus padrões de expressão são diferentes. "
p Os co-autores principais e bolsistas de pós-doutorado de Rice, Michele Di Pierro e Ryan Cheng, usaram dados de sequenciamento ChIP para uma célula de linfoblasto humano que sondas 84 proteínas de ligação a DNA diferentes e 11 modificações químicas de histonas. As proteínas histonas ajudam a organizar o genoma, agindo como carretéis em torno dos quais o DNA se enrola.
p Usando dados de apenas alguns dos cromossomos, eles treinaram uma rede neural personalizada chamada MEGABASE (anotação genômica de entropia máxima de biomarcadores associados a conjuntos estruturais) para gerar uma sequência de tipos de cromatina. Isso revelou como as marcas epigenéticas estavam relacionadas aos compartimentos, eles disseram. Uma vez treinado, eles validaram o modelo MEGABASE alimentando-o com dados dos cromossomos restantes. Isso produziu um novo conjunto de tipos estruturais para análise pelo programa MiChroM da equipe do Rice, um primo do algoritmo de paisagem de energia AWSEM do laboratório, que prevê as estruturas das proteínas. O algoritmo MiChroM previu as estruturas 3-D dos cromossomos.
p "Nossos resultados apóiam a ideia de que a compartimentação nos cromossomos surge da separação de fases de diferentes tipos de cromatina no núcleo, como a separação de óleo e água, "Cheng disse.
p Quando os pesquisadores reduziram o conjunto de dados original para apenas 11 marcações de histonas e executaram os cálculos novamente, os resultados foram apenas ligeiramente diferentes. Em última análise, eles determinaram que os dados das histonas por si só são suficientes para prever a forma de um cromossomo. "Há um código bem definido que conecta as marcações das histonas à estrutura, "Di Pierro disse." Está bem conservado, portanto, é provável que tenha uma função. "
p Para validar sua teoria, os pesquisadores compararam seus resultados com mapas de contato de linfoblastos gerados por Hi-C. Esta técnica experimental, que usa sequenciamento de alto rendimento para identificar padrões de dobramento no DNA, foi desenvolvido pelo co-autor Erez Lieberman Aiden, diretor do Centro de Arquitetura do Genoma de Baylor e pesquisador sênior do CTBP.
p "Este artigo diz que podemos pegar informações unidimensionais sobre histonas e usá-las com nossas ferramentas de big data para prever a estrutura tridimensional, "Wolynes disse.
p Seu sucesso aproxima a equipe do objetivo final de uma teoria que prevê a arquitetura de um genoma inteiro. Contudo, um problema do ovo ou da galinha permanece:a cromatina se dobra por causa dos marcadores, ou os marcadores aparecem por causa da dobra?
p "É tudo parte do nosso fascínio pela forma como a vida funciona, "Di Pierro disse." É um belo problema. "














