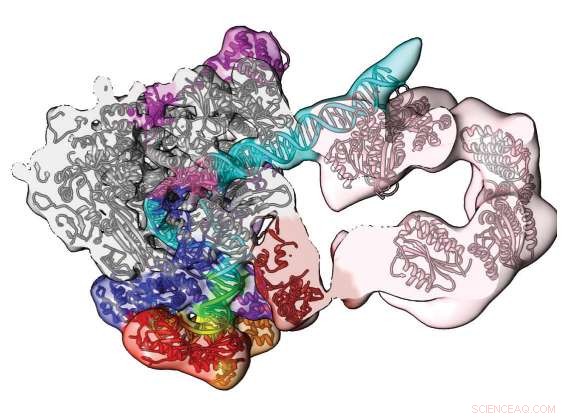
 p A supercomputação e a microscopia crioeletrônica revelam esta seção do complexo de pré-iniciação humana. O mapa e o modelo de densidade de conformação aberta mostram o caminho do DNA (azul / verde) e seu envolvimento pelo componente do fator de transcrição TFIIH (rosa). Reproduzido com permissão da Macmillan Publishers Ltd:He, Y. et al. Visualização de resolução quase atômica da abertura do promotor da transcrição humana. Natureza 533, 359–365 (2016).
p A supercomputação e a microscopia crioeletrônica revelam esta seção do complexo de pré-iniciação humana. O mapa e o modelo de densidade de conformação aberta mostram o caminho do DNA (azul / verde) e seu envolvimento pelo componente do fator de transcrição TFIIH (rosa). Reproduzido com permissão da Macmillan Publishers Ltd:He, Y. et al. Visualização de resolução quase atômica da abertura do promotor da transcrição humana. Natureza 533, 359–365 (2016).
p Parece algo saído dos borgs de Jornada nas estrelas. Robôs nanodimensionados se montam para formar máquinas biológicas que fazem o trabalho que o mantém vivo. E, no entanto, algo assim realmente acontece. p Cada célula do nosso corpo - sejam de carne e sangue, cérebro e tudo mais - tem DNA idêntico, a escada torcida de ácidos nucléicos codificados exclusivamente para cada organismo. Assembléias complexas que se assemelham a máquinas moleculares pegam pedaços de DNA chamados genes e fazem uma célula cerebral quando necessário, ao invés de, dizer, uma célula óssea. Essas máquinas moleculares são tão complexas, ainda tão pequeno, que os cientistas hoje estão apenas começando a entender sua estrutura e função usando os mais recentes microscópios e supercomputadores. Máquinas moleculares biológicas podem estabelecer a base para o desenvolvimento de curas para doenças como o câncer. Quão pequeno se pode ver, e o que encontraremos?
p A microscopia crioeletrônica combinada com simulações de supercomputador criaram o melhor modelo até agora, com detalhes próximos ao nível atômico, de uma máquina molecular vital, o complexo de pré-iniciação humana (PIC). Uma equipe de ciência da Northwestern University, Laboratório Nacional de Berkeley, Georgia State University, e UC Berkeley publicou seus resultados no PIC de maio de 2016 na revista
Natureza .
p "Pela primeira vez, foram detalhadas as estruturas dos grupos complexos de moléculas que abrem o DNA humano, "disse o co-autor do estudo Ivaylo Ivanov, professor associado de química da Georgia State University. Ivanov liderou o trabalho computacional que modelou os átomos das diferentes proteínas que atuam como engrenagens da máquina molecular PIC.
p O PIC encontra genes associados à produção de uma proteína específica, tal como um anticorpo ou uma enzima. Lá, o PIC separa as duas fitas de DNA e alimenta a fita codificadora para a enzima RNA polimerase II. Isso inicia a transcrição, onde os bits de DNA são copiados pela RNA polimerase II em uma única fita de RNA mensageiro. O RNA segue seu caminho até as "fábricas de proteínas" na célula, chamadas de ribossomos, que os tomam como ordens para as proteínas a serem produzidas. Se o DNA é como o projeto de uma nova casa, Os RNAs são instruções para os "contratados" na estação de trabalho do ribossomo. As proteínas fabricadas são como as unhas, Madeira, gesso, e quase tudo na casa.
p A experiência começou com imagens meticulosamente tiradas do PIC. Eles foram feitos por um grupo liderado pela co-autora do estudo Eva Nogales, professor do Departamento de Biologia Molecular e Celular da UC Berkeley e também Cientista Sênior do Laboratório Nacional Lawrence Berkeley e Howard Hughes Medical Investigator.
p O grupo de Nogales usou microscopia crioeletrônica (crio-EM), uma estrela em ascensão em técnicas de laboratório. Eles congelaram criogenicamente o PIC humano ligado ao DNA. O congelamento o manteve em um quimicamente ativo, ambiente quase natural. Em seguida, eles o eletrocutaram com feixes de elétrons. Graças aos avanços recentes na tecnologia de detector de elétrons direto, O cryo-EM agora pode gerar imagens com resolução quase atômica de estruturas biológicas grandes e complicadas que se mostraram muito difíceis de cristalizar. A técnica go-to, Cristalografia de raio-x, requer espécimes cristalizados, e o cryo-EM evita essa etapa difícil.
p Mais de 1,4 milhão de cryo-EM 'freeze frames' de PIC foram processados usando supercomputadores no Centro Nacional de Pesquisa de Energia para Computação Científica para filtrar o ruído de fundo e reconstruir mapas de densidade tridimensionais que mostram detalhes na forma da molécula que nunca existiu visto antes.
p "O Cryo-EM está passando por uma grande expansão, assim como todos os softwares de computador usados para gerar os mapas de densidade e também para interpretá-los como fizemos neste estudo, "Nogales disse." Isso nos permite obter uma resolução mais alta de mais estruturas em diferentes estados, para que possamos descrever não apenas uma imagem de sua aparência, mas várias fotos mostrando como eles estão se movendo. Não vemos um continuum, mas vemos instantâneos através do processo de ação. "
p Em seguida, os cientistas do estudo construíram um modelo preciso que deu sentido físico aos mapas de densidade de PIC usando XSEDE, o eXtream Science and Engineering Discovery Environment, financiado pela National Science Foundation. O XSEDE permite que os cientistas compartilhem recursos de computação de forma interativa, dados e experiência por meio de um único sistema virtual. A equipe de Ivaylo Ivanov executou mais de quatro milhões de horas básicas de simulações no supercomputador Stampede no Texas Advanced Computing Center para modelar máquinas moleculares complexas, incluindo aqueles para este estudo. O trabalho mais amplo da máquina molecular de Ivanov também inclui uma alocação XSEDE de 1,7 milhões de horas centrais no supercomputador Comet no San Diego Supercomputing Center.
p "Tenho usado os recursos do XSEDE há mais de 12 anos, "Ivanov disse." Sem a disponibilidade de recursos XSEDE, todas as nossas pesquisas teriam sido muito mais limitadas em termos dos sistemas que podemos abordar. Para nós, O XSEDE foi absolutamente essencial. "
p O objetivo de todo esse esforço computacional é produzir modelos atômicos que contem a história completa da estrutura e função do complexo de moléculas de proteínas. Para chegar lá, a equipe de Ivanov pegou os doze componentes da montagem PIC e criou modelos de homologia para cada componente que explicava suas sequências de aminoácidos e sua relação com estruturas semelhantes de proteínas 3-D conhecidas.
p Em seguida, eles aproximaram as densidades experimentais que a equipe de Nogales encontrou em uma grade. "Podemos usar um método chamado ajuste flexível de dinâmica molecular, "explicou Ivanov, "onde você essencialmente executa uma simulação de dinâmica molecular. E você usa a densidade experimental para desviar os átomos na simulação de dinâmica molecular para se moverem para as regiões mais densas do mapa EM. Esse é o processo de adaptação flexível ao mapa EM."
p Eles refinaram o modelo com o pacote de refinamento cristalográfico Phoenix. "Essa é uma técnica complementar que nos permite posicionar as cadeias laterais e melhorar o modelo para que possamos capturar todos os detalhes que estão presentes no mapa de densidade, "Ivanov disse.
p XSEDE era "absolutamente necessário" para esta modelagem, disse Ivanov. "Quando incluímos água e contra-íons, além do complexo PIC em uma caixa de simulação de dinâmica molecular, obtemos o tamanho do sistema de simulação de mais de um milhão de átomos. Não se pode executar isso em uma estação de trabalho ou mesmo em um modesto cluster. Para isso, realmente precisamos ir a mil núcleos. Nesse caso, subimos para dois mil e quarenta e oito núcleos. E para isso precisávamos de acesso ao Stampede, "Ivanov disse.
p Um dos insights obtidos no estudo é um modelo de trabalho de como o PIC abre a dupla hélice de DNA estável para transcrição. Nogales explicou que era possível imaginar uma corda feita de dois fios enrolados um no outro. Segure uma das pontas com muita força. Pegue a outra e torça-a na direção oposta da linha para desenrolar o cordão. É basicamente assim que as máquinas vivas que nos mantêm vivos fazem.
p "O DNA precisa ser aberto e movido para o sítio ativo da polimerase para codificar o primeiro nucleotídeo do RNA, "explicou Nogales." O complexo de pré-iniciação está mantendo as duas fitas do DNA juntas em uma extremidade, de modo que eles não podem se mover e eles não podem abrir. Do outro lado do PIC há uma máquina que usa energia para empurrar o DNA, torcendo-o na direção oposta em que os dois fios são enroscados. E quando isso acontecer, entre os dois lados, os fios se abrirão, "disse Nogales.
p Este estudo resolveu a estrutura dessa máquina molecular que atua como dedos torcendo, o componente do fator de transcrição TFIIH. "TFIIH tem uma subunidade de translocase, cujo papel é empurrar simultaneamente o DNA em direção ao sítio ativo da polimerase e desenrolar o DNA. Pela combinação de empurrar e desenrolar, efetivamente você está separando as duas fitas do DNA, "Ivanov disse.
p Ambos os cientistas disseram que estão apenas começando a obter uma compreensão da transcrição em nível atômico, crucial para a expressão do gene e, em última análise, a doença. "Muitos estados de doença surgem porque há erros em quanto um determinado gene está sendo lido e quanto uma determinada proteína com certa atividade na célula está presente, "Nogales disse." Esses estados de doença podem ser devido ao excesso de produção da proteína, ou, inversamente, não o suficiente. É muito importante entender o processo molecular que regula essa produção para que possamos entender o estado da doença. ”
p "Este trabalho ilustra bem dois princípios gerais que guiarão a ciência nos próximos anos, "comentou Peter Preusch, oficial do programa com o National Institutes of Health (NIH). "Um é a aplicação de métodos híbridos - combinações de métodos biofísicos, incluindo cristalografia de raios-X e crioEM, juntamente com métodos computacionais de grande escala para integrar informações sobre complexos moleculares maiores. Dois, há a necessidade de que a ciência da equipe extraia a experiência de vários investigadores para resolver problemas que não podem ser resolvidos por um único laboratório trabalhando sozinho. "Peter Preusch é o chefe do setor de biofísica, Divisão de Biologia Celular e Biofísica, Instituto Nacional de Ciências Médicas Gerais, NIH.
p Embora este trabalho fundamental não produza curas diretamente, estabelece a base para ajudar a desenvolvê-los no futuro, disse Ivanov. "Para entender a doença, temos que entender como esses complexos funcionam em primeiro lugar ... Uma colaboração entre modeladores computacionais e biólogos estruturais experimentais pode ser muito frutífera no futuro. "
p O estudo de artigos da natureza de maio de 2016 (DOI:10.1038 / nature17970), "Visualização de resolução quase atômica da abertura do promotor da transcrição humana, "foi de autoria de Yuan He, Lawrence Berkeley National Laboratory e agora na Northwestern University; Chunli Yan e Ivaylo Ivanov, Georgia State University; Jie Fang, Carla Inouye, Robert Tjian, Eva Nogales, UC Berkeley. O financiamento veio do Instituto Nacional de Ciências Médicas Gerais (NIH) e da National Science Foundation.