Certos tipos de moléculas formam padrões quando depositados em substratos. Dispositivos fotovoltaicos e sensores de compostos orgânicos dependem desse fenômeno de auto-organização. Físicos da Ludwig-Maximilians-Universitaet em Munique, Alemanha, desenvolveram agora um modelo que prevê esses padrões e, assim, permite a otimização da síntese molecular no futuro.
Algumas classes de moléculas são capazes de se organizar em padrões específicos nas superfícies. Essa capacidade de se auto-organizar é crucial para muitas aplicações tecnológicas, que dependem da montagem de estruturas ordenadas em superfícies. Contudo, até agora tem sido virtualmente impossível prever ou controlar o resultado de tais processos.
Agora, um grupo de pesquisadores liderado pela Dra. Bianca Hermann, um físico do Centro de Nanociência (CeNS) da LMU Munique, relata um avanço significativo:Ao combinar física estatística e simulações detalhadas com imagens obtidas por microscopia de tunelamento de varredura (STM), a equipe foi capaz de formular um modelo simples que pode prever os padrões observados. “Com a ajuda da modelo, podemos gerar uma grande variedade de padrões que reproduzem surpreendentemente bem os arranjos observados experimentalmente ", diz Hermann. "Queremos estender essa abordagem a outras simetrias de superfície. Já agora as áreas de eletrônica molecular, aplicações de sensores, a catálise de superfície e a fotovoltaica orgânica podem lucrar com nosso modelo. Sua capacidade de prever estruturas formadas por auto-organização permite a otimização de blocos de construção moleculares antes da síntese. "( Nano Letras conectados, 16 de fevereiro de 2010)
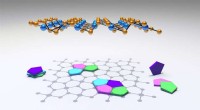
Quando a "mãe natureza" faz a engenharia, moléculas podem se auto-organizar em estruturas complexas - uma primeira etapa na formação de membranas, células e outros sistemas moleculares. O princípio da auto-organização, o que permite o uso muito econômico de recursos, também é explorado na produção de superfícies funcionalizadas necessárias em eletrônica molecular, aplicações de sensores, catálise e componentes fotovoltaicos. A ideia do processo de fabricação é que os componentes moleculares sejam colocados em contato com um material substrato, e então "magicamente" encontre suas posições preferidas na rede molecular desejada. Os componentes iniciais são selecionados para exibir características estruturais e químicas específicas para a aplicação prevista. Contudo, a otimização dos adlayers moleculares depende em grande parte de uma abordagem de tentativa e erro, e, portanto, é complicado e demorado.
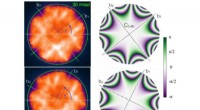
Para desenvolver o novo modelo de local de interação molecular, O grupo do Dr. Herrmann colaborou com Priv. Doz. Dr. Thomas Franosch e Professor Erwin Frey dentro do Cluster of Excellence "Nanosystems Initiative Munich" (NIM). O problema foi resolvido usando uma abordagem da física estatística conhecida como método de Monte Carlo, que permite realizar uma simulação de computador detalhada sobre as estatísticas de interações moleculares. Os motivos estruturais assim gerados foram comparados com imagens experimentais de alta resolução de padrões moleculares obtidos por STM. Marta Balbás Gambra, um estudante de doutorado, começou cada simulação com uma representação matemática de uma coleção de centenas de partículas orientadas aleatoriamente de conformação definida. Essas moléculas esquemáticas foram então perturbadas - computacionalmente - adicionando energia, fazendo com que a população adote uma nova configuração.
Usando esta estratégia de simulação, pode-se gerar uma maior variedade de padrões do que os encontrados naturalmente, e muitos deles correspondiam intimamente aos padrões moleculares reais revelados pelo STM. "Em um caso, previmos um padrão que só foi verificado mais tarde com o STM", relata o aluno de doutorado Carsten Rohr. De acordo com as leis da termodinâmica, os sistemas físicos tendem a adotar o estado com a energia mais favorável (ou seja, mais baixa). Testes experimentais mostraram que diferentes configurações moleculares se convertem até que predomine um arranjo que lembra marcas de pneus. E realmente, a abordagem de Monte Carlo previu que esse arranjo corresponde ao estado com a energia mais baixa.
"No fim, fomos capazes de mostrar que a geometria molecular e algumas características salientes codificam os motivos estruturais observados ", explica o teórico Franosch. "Pretendemos estender a abordagem a outros tipos de simetrias de superfície, mas o modelo já fornece uma ferramenta teórica importante, porque nos ajuda a prever o tipo de padrão de superfície que uma dada molécula funcional formará. Isso significa que o design das moléculas pode ser otimizado durante a fase sintética, de modo a obter superfícies com as características desejadas ", diz Hermann. Os físicos do grupo, que vêm de diferentes origens científicas e foram capazes de reunir seus conhecimentos para este projeto, prever múltiplas aplicações potenciais para seu modelo em eletrônica molecular, tecnologia de sensor, catálise e fotovoltaica. Outras possibilidades incluem seu uso para prever os resultados de outros tipos de interações moleculares também em substratos parcialmente padronizados.