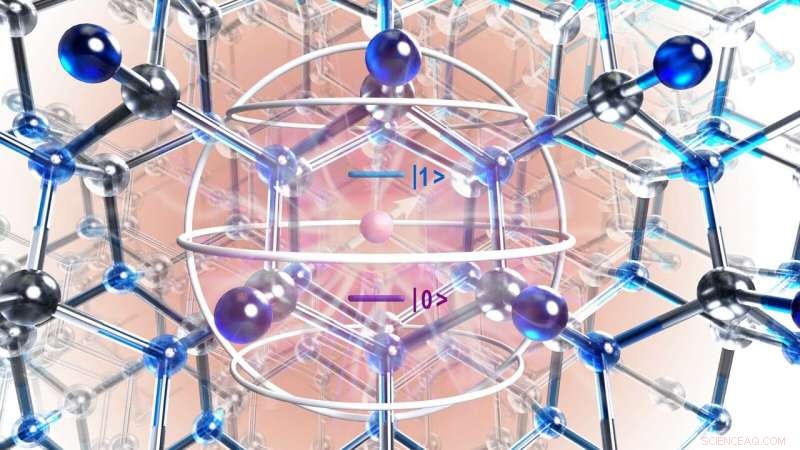
Renderização artística da estrutura atômica do cristal de carboneto de silício mostrando defeito (círculo roxo) e região de interesse identificada com a teoria da mecânica quântica (esfera de prata). Crédito:Universidade de Chicago
Os computadores quânticos têm um potencial enorme para cálculos usando algoritmos novos e envolvendo quantidades de dados muito além da capacidade dos supercomputadores de hoje. Embora esses computadores tenham sido construídos, eles ainda estão em sua infância e têm aplicabilidade limitada para resolver problemas complexos em ciência de materiais e química. Por exemplo, eles apenas permitem a simulação das propriedades de alguns átomos para pesquisa de materiais.
Cientistas do Laboratório Nacional de Argonne do Departamento de Energia dos EUA (DOE) e da Universidade de Chicago (UChicago) desenvolveram um método que abre caminho para o uso de computadores quânticos para simular moléculas realistas e materiais complexos, cuja descrição requer centenas de átomos.
A equipe de pesquisa é liderada por Giulia Galli, diretor do Centro Integrado de Materiais Computacionais do Meio-Oeste (MICCoM), líder de grupo na divisão de Ciência de Materiais de Argonne e membro do Centro de Engenharia Molecular em Argonne. Galli também é Professor de Estrutura Eletrônica e Simulações da Família Liew na Escola Pritzker de Engenharia Molecular e Professor de Química na UChicago. Ela trabalhou neste projeto com o cientista assistente Marco Govoni e o estudante de graduação He Ma, ambas parte da divisão de Ciência de Materiais de Argonne e UChicago.
"Nosso método de cálculo recém-desenvolvido, "Galli disse, "melhora muito a precisão alcançável com os métodos de mecânica quântica existentes em relação aos cálculos para defeitos específicos em materiais cristalinos, e o implementamos em um computador quântico. "
Nas últimas três décadas, abordagens teóricas da mecânica quântica têm desempenhado um papel importante na previsão das propriedades de materiais relevantes para a ciência da informação quântica e materiais funcionais para aplicações de energia, abrangendo catalisadores e sistemas de armazenamento de energia. Contudo, essas abordagens são computacionalmente exigentes, e ainda é um desafio aplicá-los a complexos, materiais heterogêneos.
"Em nossa pesquisa, desenvolvemos uma teoria de incorporação quântica que permitiu a simulação de 'defeitos de spin' em sólidos por acoplamento de hardware de computação quântica e clássica, "Disse Govoni. Esses tipos de defeitos em sólidos têm aplicabilidade para o desenvolvimento de materiais para processamento de informações quânticas e aplicações de detecção em nanoescala muito além das capacidades atuais.
"A nossa é uma estratégia avançada e poderosa na ciência de materiais computacionais com o potencial de prever as propriedades de materiais complexos com mais precisão do que os métodos atuais mais avançados podem fazer no momento, "Govoni acrescentou.
A equipe testou primeiro o método de incorporação quântica em um computador clássico, aplicando-o aos cálculos das propriedades dos defeitos de rotação em diamante e carboneto de silício. "Os pesquisadores anteriores estudaram extensivamente os defeitos no diamante e no carboneto de silício, portanto, tínhamos dados experimentais abundantes para comparar com as previsões do nosso método, "disse Ma. A boa concordância entre teoria e experimento deu à equipe confiança na confiabilidade de seu método.
A equipe então passou a testar os mesmos cálculos em um simulador quântico e, finalmente, no computador quântico IBM Q5 Yorktown. Os resultados confirmaram a alta precisão e eficácia de seu método de incorporação quântica, estabelecendo um trampolim para resolver muitos tipos diferentes de problemas da ciência dos materiais em um computador quântico.
Galli notou que, "Com a inevitável maturidade dos computadores quânticos, esperamos que nossa abordagem seja aplicável à simulação de regiões de interesse em moléculas e materiais para a compreensão e descoberta de catalisadores e novas drogas, bem como soluções aquosas contendo espécies complexas dissolvidas. "
A equipe de Galli faz parte do MICCoM, com sede em Argonne; o Chicago Quantum Exchange, com sede em UChicago; e o projeto QISpin financiado pelo Escritório de Pesquisa Científica da Força Aérea.
Sua pesquisa aproveitou o software WEST desenvolvido dentro do MICCoM e fez uso de vários recursos de computação além do computador quântico IBM disponível ao público:o Argonne Leadership Computing Facility e o National Energy Research Scientific Computing Center, ambos DOE Office of Science User Facilities; e o Centro de Pesquisa de Computação da Universidade de Chicago.
O trabalho da equipe é apresentado em um artigo intitulado "Simulações quânticas de materiais em computadores quânticos de curto prazo", que aparece na edição de julho de 2020 da npj materiais computacionais .