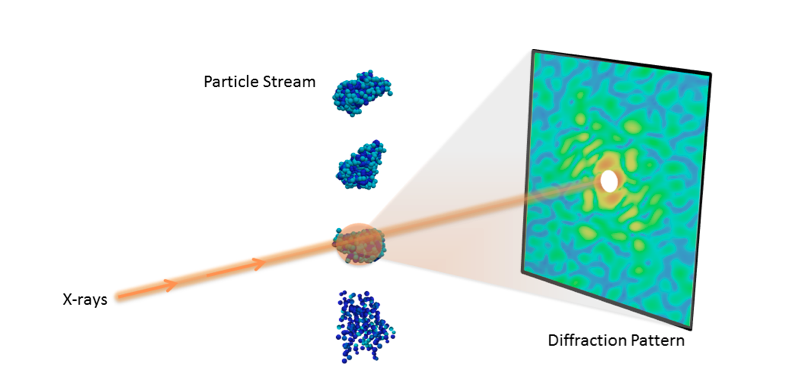
 p Configuração experimental para um experimento de difração de partícula única. Crédito:Peter Zwart, Berkeley Lab
p Configuração experimental para um experimento de difração de partícula única. Crédito:Peter Zwart, Berkeley Lab
p Compreender a estrutura molecular 3D de nanoobjetos importantes, como proteínas e vírus, é crucial na biologia e na medicina. Com os avanços recentes na tecnologia de raios-X, os cientistas agora podem coletar imagens de difração de partículas individuais, em última análise, permitindo que os pesquisadores visualizem as moléculas em temperatura ambiente. p Contudo, determinar a estrutura 3D a partir desses experimentos de difração de partícula única é um obstáculo significativo. Por exemplo, as taxas de aquisição de dados atuais são muito limitantes, normalmente resultando em menos de 10 instantâneos úteis por minuto, limitando a quantidade de recursos que podem ser resolvidos. Adicionalmente, as imagens são frequentemente altamente corrompidas com ruído e outros artefatos experimentais, dificultando a interpretação adequada dos dados.
p Para enfrentar esses desafios, uma equipe de pesquisadores do Lawrence Berkeley National Laboratory (Berkeley Lab) desenvolveu uma nova estrutura algorítmica chamada de faseamento iterativo multicamadas (M-TIP) que utiliza técnicas matemáticas avançadas para determinar a estrutura molecular 3D a partir de conjuntos muito esparsos de ruído, dados de partícula única. Essa abordagem permite essencialmente que os pesquisadores extraiam mais informações de experimentos com dados limitados. Os matemáticos aplicados Jeffrey Donatelli e James Sethian, e o biocientista físico Peter Zwart introduziu essa estrutura expandindo um algoritmo que eles desenvolveram originalmente para resolver a reconstrução de um experimento de espalhamento de raios-X relacionado, chamado de espalhamento de raios-X por flutuação. Um artigo que descreve a estrutura M-TIP foi publicado em 26 de junho no
Anais da Academia Nacional de Ciências .
p "Esta abordagem tem o potencial de revolucionar o campo, "diz Zwart." Dado que é difícil obter muitos dados bons, abordagens que reduzem a quantidade de dados necessários para obter imagens nanoobjetos 3D com sucesso devem receber uma recepção calorosa. "
p Donatelli, Sethian e Zwart fazem parte do CAMERA (Centro de Matemática Avançada para Aplicações de Pesquisa de Energia), cuja missão é criar a matemática de ponta necessária para lidar com dados de muitas das instalações científicas mais avançadas do DOE. CAMERA é financiado conjuntamente pelos programas Advanced Scientific Computing Research e Basic Energy Sciences no Office of Science do DOE.
p
Difração de partícula única
p O recente advento dos lasers de elétrons livres de raios-X (XFELs) possibilitou várias novas técnicas experimentais para estudar biomoléculas que eram inviáveis com fontes de luz tradicionais. Uma dessas técnicas é a difração de partícula única, que coleta um grande número de instantâneos de difração de raios-X com apenas uma única partícula no feixe. Aproveitando o poder extremo dos XFELs, os pesquisadores podem coletar sinais mensuráveis até mesmo das partículas mais ínfimas.
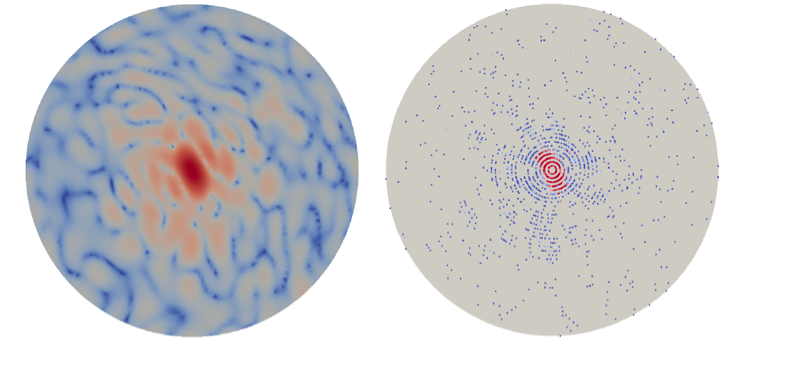 p Um exemplo de imagem de difração de partícula única limpa (esquerda) e a mesma imagem de difração após contaminação por ruído (direita). Crédito:Peter Zwart, Berkeley Lab
p Um exemplo de imagem de difração de partícula única limpa (esquerda) e a mesma imagem de difração após contaminação por ruído (direita). Crédito:Peter Zwart, Berkeley Lab
p Uma grande vantagem oferecida por esta técnica de difração de partícula única é a capacidade de estudar como diferentes cópias de uma molécula variam ou mudam de forma. Uma vez que cada imagem vem de uma única partícula, essas variações podem ser capturadas no experimento, em contraste com os métodos de imagem tradicionais, como cristalografia ou espalhamento de raios-X de pequeno ângulo, onde os pesquisadores só podem medir uma média sobre todos os diferentes estados da amostra molecular.
p Contudo, determinar a estrutura 3D a partir de dados de difração de partícula única é um desafio. Começar, quando cada partícula é fotografada, sua orientação é desconhecida e precisa ser recuperada para combinar corretamente os dados em um volume de difração 3D. Este problema é agravado se a molécula pode assumir diferentes formas, o que requer classificação adicional das imagens. Além disso, as informações de fase não são registradas nas imagens de difração e devem ser recuperadas para concluir a reconstrução. Finalmente, mesmo com XFELs poderosos, o número de fótons espalhados é muito pequeno, resultando em imagens extremamente barulhentas, que pode ser ainda mais contaminado por problemas sistemáticos de fundo e de leitura do detector.
p As abordagens anteriores são baseadas na resolução do problema de reconstrução em etapas separadas, onde cada problema individual é tratado separadamente. Infelizmente, uma desvantagem dessas abordagens em série é que elas não alavancam facilmente características conhecidas anteriores sobre a aparência da molécula. Além disso, qualquer erro cometido em uma etapa é propagado para a próxima, resultando em um aumento adicional no erro. Essa "bola de neve do erro" acaba por degradar a qualidade da reconstrução obtida na etapa final.
p
O melhor de dois mundos
p Em vez de resolver os problemas computacionais em etapas separadas, o algoritmo M-TIP da equipe resolve todas as partes do problema simultaneamente. Esta abordagem aproveita informações prévias sobre a estrutura para reduzir significativamente os graus de liberdade do problema em todas as etapas, e, conseqüentemente, reduzir as informações necessárias para conseguir uma reconstrução 3D.
p "As técnicas de otimização de caixa preta padrão podem incorporar conhecimento prévio na reconstrução, mas descartar toda a estrutura do problema, ao passo que resolvê-lo em subetapas seriais completamente separadas explora a estrutura do problema, mas joga fora quase todas as informações anteriores sobre como a solução pode ser, "Donatelli disse." M-TIP aproveita o melhor dos dois mundos, explorando a estrutura do problema para quebrar a computação em vários pedaços gerenciáveis e, em seguida, refinar iterativamente sobre todos esses pedaços para chegar a uma solução que seja consistente com ambos os dados e quaisquer restrições estruturais. "
p Usando esta técnica, a equipe foi capaz de determinar a estrutura 3D a partir de contagens de imagens extremamente baixas a partir de dados simulados, de 6 a 24 imagens para dados sem ruído e 192 imagens de dados altamente contaminados.
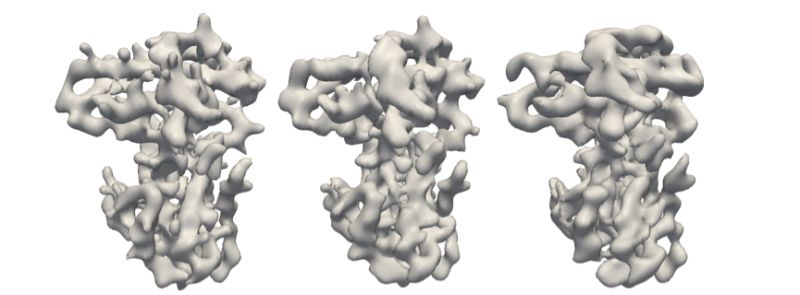 p Proteína de retinoblastoma original (esquerda) e reconstruções usando o algoritmo M-TIP com 24 imagens limpas (meio) e 192 imagens com ruído (direita), como mostrado na Figura 2. Crédito:Peter Zwart, Berkeley Lab
p Proteína de retinoblastoma original (esquerda) e reconstruções usando o algoritmo M-TIP com 24 imagens limpas (meio) e 192 imagens com ruído (direita), como mostrado na Figura 2. Crédito:Peter Zwart, Berkeley Lab
p
Abrindo novos caminhos
p Este trabalho faz parte de uma nova iniciativa de colaboração entre SLAC National Accelerator Laboratory, CÂMERA, o Centro de Computação Científica de Pesquisa Energética Nacional (NERSC) e o Laboratório Nacional de Los Alamos como parte do Projeto de Computação Exascale do DOE (ECP). O objetivo do projeto é desenvolver as ferramentas computacionais necessárias para realizar a análise de dados em tempo real de experimentos que estão sendo conduzidos no Linac Coherent Light Source (LCLS) do SLAC. Com atualizações para a linha de luz, LCLS-II planeja gerar vários terabytes de dados por segundo, que, por exemplo, permitirá que os cientistas expandam bastante os experimentos atuais de uma única partícula. A análise de todos esses dados em tempo real exigirá novos algoritmos e grandes máquinas de computação. O algoritmo M-TIP servirá como parte desse processo.
p "Esses são alguns dos problemas mais desafiadores da ciência de dados computacionais, "diz Sethian." Para enfrentá-los, precisamos explorar uma variedade de tecnologias, incluindo arquiteturas de computação exascale emergentes, redes sofisticadas de alta velocidade, e os algoritmos matemáticos mais avançados disponíveis. Reunir cientistas do CAMERA com projetos de aplicação exascale abriu a porta para a construção de ferramentas para abordar alguns problemas urgentes em biologia e ciências dos materiais. "
p Os pesquisadores observam que esses são apenas os primeiros passos. Para que o método esteja pronto para ser implantado, outros obstáculos devem ser superados.
p "A ciência experimental é confusa, "diz Zwart." Existem efeitos experimentais adicionais que devem ser levados em consideração para que possamos obter os melhores resultados possíveis. "
p "Felizmente, M-TIP é uma técnica muito modular, "acrescenta Donatelli, "tão, ele é adequado para modelar muitos desses efeitos adicionais sem a necessidade de alterar a estrutura algorítmica principal. "
p A equipe está atualmente trabalhando no estudo desses efeitos como parte da Single Particle Initiative, um grande, colaboração multi-institucional dedicada a abordar questões teóricas e práticas em imagem de molécula única baseada em X-FEL, em última análise, levando a fornecer à comunidade científica as ferramentas necessárias para abrir novos caminhos na biologia, medicina e ciências da energia.