Pepsi-SAXS:Novo método de análise de proteínas 50 vezes mais rápido do que os análogos. Crédito:MIPT
Pepsi-SAXS é um novo, método altamente eficiente para cálculo de perfis de espalhamento de raios-X, que são necessários para a análise da molécula de proteína no estado de solução. O método foi criado por cientistas da Université Grenoble Alpes e MIPT, liderado por Sergei Grudinin. A equipe testou seu método, e os resultados foram publicados pela International Union of Crystallography em seu jornal Acta Crystallographica Seção D:Biologia Estrutural .
As proteínas têm estrutura complexa e tamanho extremamente pequeno, na ordem de vários nanômetros. Para estudá-los, os pesquisadores precisam encontrar métodos incomuns, porque as amostras de proteínas são facilmente destruídas e suas propriedades alteradas em experimentos. O conhecimento das estruturas e mecanismos funcionais das biomoléculas permite que novos medicamentos sejam desenvolvidos não por tentativa e erro - tecnicamente chamados de triagem de alto rendimento - mas de uma forma mais focada.
Uma das técnicas utilizadas para estudar proteínas é a análise de raios X espalhados por elas. Os pesquisadores precisam usar raios-X e não luz comum para ampliar átomos individuais com um tamanho característico da ordem de 0,1 nanômetro. Quanto menor o objeto, quanto menor o comprimento de onda da luz que deve ser usada para observá-lo. A luz visível compreende comprimentos de onda entre 400 e 700 nanômetros. Raios X, por outro lado, têm um comprimento de onda muito menor e, portanto, podem ser usados para examinar estruturas moleculares.
"O novo método nos permite traçar curvas de espalhamento com eficiência e precisão, e analisar a estrutura tridimensional de uma amostra, "diz a aluna do MIPT Maria Garkavenko, um co-autor do artigo. "Entre outras coisas, Pepsi-SAXS aumenta a eficiência da modelagem e a precisão da previsão da estrutura da macromolécula tridimensional. "
Espalhamento de raios-X de pequeno ângulo, ou SAXS, é uma técnica experimental que envolve o espalhamento de raios X de uma amostra e sua coleta em ângulos muito pequenos. Como resultado, é obtido um gráfico da intensidade do feixe de raios-X espalhado em função do ângulo de incidência. Usando este gráfico, uma amostra de proteína pode ser comparada a outras amostras no banco de dados experimental para determinar sua estrutura e propriedades.
Comparado com outras técnicas usadas para determinar a estrutura da amostra, SAXS é muito mais simples e barato. Requer apenas uma preparação mínima de amostra, e as proteínas não precisam ser congeladas ou cristalizadas. As amostras são estudadas em solução e em seu estado funcional. Isso torna os resultados muito mais confiáveis, porque a preparação da amostra às vezes pode alterar o estado e as propriedades de uma proteína. Outra vantagem importante do método é que ele não é destrutivo, o que significa que a amostra experimental permanece praticamente inalterada pelos raios-X.
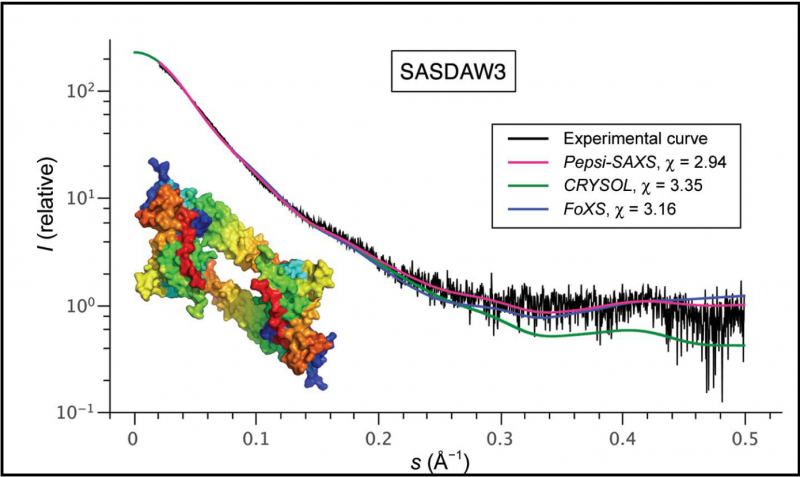
A Figura 1 mostra os resultados de uma série de experimentos, que comparou Pepsi-SAXS com dois dos métodos computacionais usados atualmente, aplicando-os à mesma amostra (SASDAW3) do banco de dados SASBDB. A intensidade média de espalhamento é representada graficamente como uma função do ângulo de espalhamento. O erro χ² do modelo computacional é o menor no caso da Pepsi-SAXS, que resulta de uma representação mais precisa da concha de hidratação. Crédito:S. Grudinin, M. Garkavenko e A. Kazennov
Mas até recentemente, SAXS tinha uma grande desvantagem:o método era computacionalmente intensivo, o que significava que não poderia ser usado se o número de experimentos fosse substancial. Demorou horas para processar os resultados de apenas um experimento. Inicialmente, o número de cálculos foi diretamente proporcional ao quadrado do número de átomos na amostra, o último número geralmente excede mil. Contudo, Na década de 1970, Heinrich Stuhrmann, um pesquisador alemão, teve uma ideia que simplificou os cálculos. Ele propôs que o espalhamento em compostos moleculares seja descrito em termos de funções de um tipo particular chamado de harmônicos esféricos. Essa abordagem provou ser um sucesso. Ao longo dos anos, uma série de ferramentas computacionais para a análise de dados SAXS foram criadas. Contribuições importantes para o seu desenvolvimento foram feitas por pesquisadores com formação científica soviética, incluindo Dmitri Svergun (atualmente trabalhando em Hamburgo), que escreveu o pacote de software ATSAS para análise de dados SAXS na pesquisa de macromoléculas biológicas. Os pesquisadores do estudo relatado aqui examinaram vários métodos computacionais e os compararam com suas próprias técnicas.
“Pepsi-SAXS significa 'expansões polinomiais de estruturas e interações de proteínas' e 'espalhamento de raios-X de baixo ângulo'. É um método adaptativo para computação rápida e precisa de perfis de espalhamento de raios-X de pequeno ângulo, "explica o aluno de doutorado do MIPT Andrei Kazennov, um co-autor do artigo. "Pepsi-SAXS pode ser adaptado ao tamanho de uma determinada amostra e à resolução dos dados experimentais."
Os pesquisadores também criaram um modelo eficiente da camada de hidratação - uma camada de moléculas de água que envolve as proteínas em solução - e o incorporaram ao software, aumentando a precisão do método.
"Nosso método foi validado em um grande conjunto de dados da BioIsis e SASBDB, os dois maiores bancos de dados biológicos, "diz Sergei Grudinin, quem orientou a pesquisa. "Mostramos que Pepsi-SAXS é cinco a 50 vezes mais rápido do que os métodos usados anteriormente, ou seja, CRYSOL, FoXS, e a técnica tridimensional de Zernike implementada no pacote SAStbx. Ao mesmo tempo, a precisão está no mesmo nível deles. "
Os pesquisadores prestaram atenção especial à análise dos resultados obtidos, que foram comparados com os dados experimentais.
A pesquisa de proteínas tem um significado fundamental para a nossa compreensão dos processos básicos subjacentes à vida, bem como para o desenvolvimento de drogas, tratamentos, e materiais orgânicos, incluindo órgãos artificiais. A nova ferramenta apresentada pelos autores pode significar um progresso 50 vezes mais rápido nessas áreas.