Acelerando a produção de novos medicamentos com aprendizado de máquina
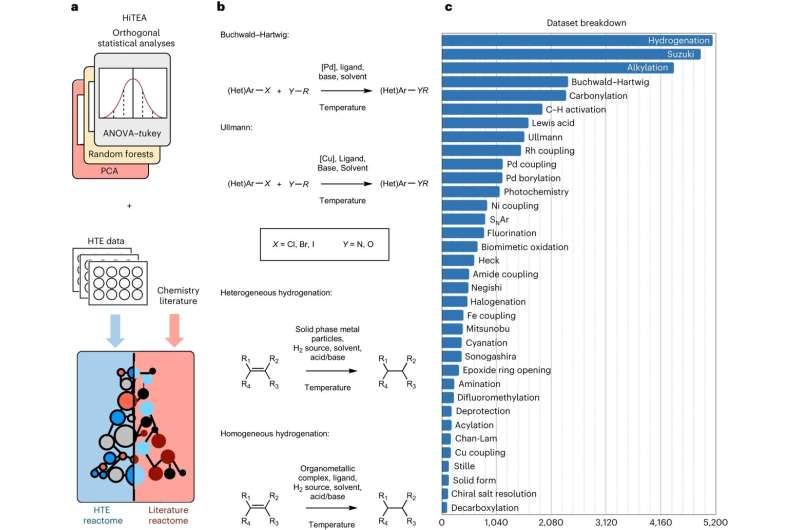
Visão geral do conjunto de dados e estrutura HTE. um , É mostrada uma visão geral do HiTEA e sua análise. A comparação do reatoma da literatura com o reatoma HiTEA revelará suporte para nossas conclusões mecanicistas (acordo de reatomas) ou revelará áreas de viés/fenômeno químico incomum (discordância de reatomas). b , São mostradas representações resumidas das quatro classes de reação analisadas pelo HiTEA nesta publicação. c , É mostrada a divisão do conjunto de dados HTE por classe de reação. Crédito:Química da Natureza (2024). DOI:10.1038/s41557-023-01393-w Os investigadores desenvolveram uma plataforma que combina experiências automatizadas com IA para prever como os produtos químicos reagirão entre si, o que poderá acelerar o processo de concepção de novos medicamentos.
Prever como as moléculas irão reagir é vital para a descoberta e fabricação de novos produtos farmacêuticos, mas historicamente este tem sido um processo de tentativa e erro, e as reações muitas vezes falham. Para prever como as moléculas reagirão, os químicos geralmente simulam elétrons e átomos em modelos simplificados, um processo que é computacionalmente caro e muitas vezes impreciso.
Agora, investigadores da Universidade de Cambridge desenvolveram uma abordagem baseada em dados, inspirada na genómica, onde experiências automatizadas são combinadas com aprendizagem automática para compreender a reatividade química, acelerando enormemente o processo. Eles chamaram sua abordagem, que foi validada em um conjunto de dados de mais de 39.000 reações farmaceuticamente relevantes, de “reactoma” químico.
Seus resultados, publicados na revista Nature Chemistry , são o produto de uma colaboração entre Cambridge e Pfizer.
“O reatoma pode mudar a forma como pensamos sobre a química orgânica”, disse a Dra. Emma King-Smith, do Laboratório Cavendish de Cambridge, a primeira autora do artigo. "Uma compreensão mais profunda da química poderia permitir-nos fabricar produtos farmacêuticos e muitos outros produtos úteis com muito mais rapidez. Mas, mais fundamentalmente, a compreensão que esperamos gerar será benéfica para qualquer pessoa que trabalhe com moléculas."
A abordagem reatoma seleciona correlações relevantes entre reagentes, reagentes e desempenho da reação a partir dos dados e aponta lacunas nos próprios dados. Os dados são gerados a partir de experimentos automatizados muito rápidos ou de alto rendimento.
"A química de alto rendimento mudou o jogo, mas acreditávamos que havia uma maneira de descobrir uma compreensão mais profunda das reações químicas do que aquela que pode ser observada a partir dos resultados iniciais de um experimento de alto rendimento", disse King-Smith.
“Nossa abordagem revela as relações ocultas entre os componentes da reação e os resultados”, disse o Dr. Alpha Lee, que liderou a pesquisa. "O conjunto de dados no qual treinamos o modelo é enorme - ajudará a levar o processo de descoberta química da tentativa e erro para a era do big data."
Em um artigo relacionado, publicado na Nature Communications , a equipe desenvolveu uma abordagem de aprendizado de máquina que permite aos químicos introduzir transformações precisas em regiões de moléculas pré-especificadas, permitindo um design de medicamentos mais rápido.
A abordagem permite que os químicos ajustem moléculas complexas – como uma mudança de design de última hora – sem ter que criá-las do zero. Fazer uma molécula no laboratório normalmente é um processo de várias etapas, como construir uma casa. Se os químicos quiserem variar o núcleo de uma molécula, a maneira convencional é reconstruí-la, como derrubar uma casa e reconstruí-la do zero. No entanto, as variações principais são importantes para o design de medicamentos.
Uma classe de reações conhecidas como reações de funcionalização de estágio final tenta introduzir transformações químicas diretamente no núcleo, evitando a necessidade de começar do zero. No entanto, é um desafio tornar a funcionalização em estágio final seletiva e controlada – normalmente há muitas regiões das moléculas que podem reagir e é difícil prever o resultado.
"As funcionalizações em estágio avançado podem produzir resultados imprevisíveis e os métodos atuais de modelagem, incluindo nossa própria intuição especializada, não são perfeitos", disse King-Smith. “Um modelo mais preditivo nos daria a oportunidade de uma melhor triagem”.
Os pesquisadores desenvolveram um modelo de aprendizado de máquina que prevê onde uma molécula reagiria e como o local da reação varia em função das diferentes condições de reação. Isso permite que os químicos encontrem maneiras de ajustar com precisão o núcleo de uma molécula.
"Pré-treinamos o modelo em um grande conjunto de dados espectroscópicos - ensinando efetivamente a química geral do modelo - antes de ajustá-lo para prever essas transformações intrincadas", disse King-Smith. Esta abordagem permitiu à equipe superar a limitação de poucos dados:há relativamente poucas reações de funcionalização em estágio final relatadas na literatura científica. A equipe validou experimentalmente o modelo em um conjunto diversificado de moléculas semelhantes a drogas e foi capaz de prever com precisão os locais de reatividade sob diferentes condições.
“A aplicação do aprendizado de máquina à química é frequentemente dificultada pelo problema de que a quantidade de dados é pequena em comparação com a vastidão do espaço químico”, disse Lee. "Nossa abordagem - projetar modelos que aprendem com grandes conjuntos de dados que são semelhantes, mas não iguais, ao problema que estamos tentando resolver - resolve esse desafio fundamental de poucos dados e pode desbloquear avanços além da funcionalização em estágio final."
Mais informações: Emma King-Smith et al, Sondando o 'reactome' químico com dados de experimentação de alto rendimento, Nature Chemistry (2024). DOI:10.1038/s41557-023-01393-w Funcionalização preditiva de minisci em estágio final com aprendizagem por transferência, Nature Communications (2024). DOI:10.1038/s41467-023-42145-1. www.nature.com/articles/s41467-023-42145-1
Informações do diário: Comunicações da Natureza , Química da Natureza