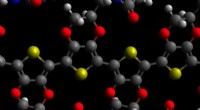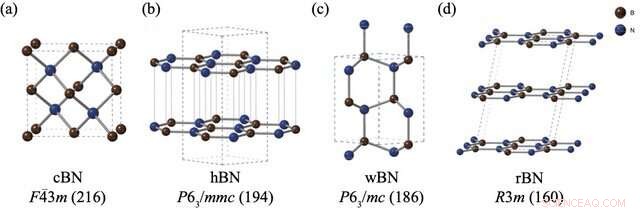
As estruturas e grupos espaciais de (a) nitreto de boro de mistura de zinco (cBN), (b) nitreto de boro hexagonal (hBN), (c) nitreto de boro wurtzita (wBN) e (d) nitreto de boro romboédrico (rBN). Os átomos de boro e nitrogênio são representados em marrom e azul, respectivamente. Crédito:Kousuke Nakano do JAIST.
O nitreto de boro (BN) é um material versátil com aplicações em diversas áreas de engenharia e ciências. Isso se deve em grande parte a uma propriedade interessante da BN chamada "polimorfismo", caracterizada pela capacidade de cristalizar em mais de um tipo de estrutura. Isso geralmente ocorre como uma resposta a mudanças de temperatura, pressão ou ambos. Além disso, as diferentes estruturas, chamadas "polimorfos", diferem notavelmente em suas propriedades físicas, apesar de terem a mesma fórmula química. Como resultado, os polimorfos desempenham um papel importante no design do material, e um conhecimento de como favorecer seletivamente a formação do polimorfo desejado é crucial nesse sentido.
No entanto, os polimorfos BN apresentam um problema particular. Apesar da realização de vários experimentos para avaliar as estabilidades relativas de polimorfos BN, não surgiu um consenso sobre este tópico. Embora os métodos computacionais sejam frequentemente a abordagem para esses problemas, os polimorfos BN representam sérios desafios às técnicas de computação padrão devido às fracas "interações de van der Waals (vdW)" entre suas camadas, o que não é contabilizado nesses cálculos. Além disso, os quatro polimorfos BN estáveis, a saber, romboédrico (rBN), hexagonal (hBN), wurtzita (wBN) e zinco-blenda (cBN), manifestam-se dentro de uma estreita faixa de energia, fazendo com que a captura de pequenas diferenças de energia juntamente com interações vdW ainda mais desafiador.
Uma equipe de pesquisa internacional liderada pelo professor assistente Kousuke Nakano do Instituto Avançado de Ciência e Tecnologia do Japão (JAIST) agora forneceu evidências para resolver o debate. Em seu estudo, eles abordaram a questão com uma estrutura de cálculos de primeiros princípios de última geração, ou seja, simulações de Monte Carlo de difusão em nó fixo (FNDMC). O FNDMC representa um passo no popular método de simulações quânticas de Monte Carlo, no qual uma "função de onda" quântica de muitos corpos parametrizada é primeiro otimizada para atingir o estado fundamental e depois fornecida ao FNDMC.
Além disso, a equipe também calculou a energia de Gibbs (o trabalho útil obtido de um sistema a pressão e temperatura constantes) de polimorfos BN para diferentes temperaturas e pressões usando a teoria do funcional da densidade (DFT) e cálculos de fônons. Este artigo foi disponibilizado on-line em 24 de março de 2022, publicado no
The Journal of Physical Chemistry C .
De acordo com os resultados do FNDMC, hBN foi a estrutura mais estável, seguida por rBN, cBN e wBN. Estes resultados foram consistentes a 0 K e 300 K (temperatura ambiente). No entanto, as estimativas DFT produziram resultados conflitantes para duas aproximações diferentes. O Dr. Nakano explica esses achados contraditórios:"Nossos resultados revelam que a estimativa de estabilidades relativas é muito influenciada pelo funcional correlacional de troca, ou a aproximação usada no cálculo DFT. Como resultado, uma conclusão quantitativa não pode ser alcançada usando os achados DFT, e uma abordagem mais precisa, como FNDMC, é necessária."
Notavelmente, os resultados do FNDMC estavam de acordo com aqueles gerados por outros métodos de computação refinados, como "acoplamento de cluster", sugerindo que o FNDMC é uma ferramenta eficaz para lidar com polimorfos, especialmente aqueles governados por forças vdW. A equipe também mostrou que pode fornecer outras informações importantes, como energias de referência confiáveis, quando os dados experimentais não estão disponíveis.
O Dr. Nakano está entusiasmado com as perspectivas futuras do método na área de ciência dos materiais. "Nosso estudo demonstra a capacidade do FNDMC de detectar pequenas mudanças de energia envolvendo forças vdW, o que estimulará o uso desse método para outros materiais de van der Waals", diz ele. "Além disso, simulações moleculares baseadas neste método preciso e confiável podem capacitar projetos de materiais, permitindo o desenvolvimento de medicamentos e catalisadores".
+ Explorar mais Aumentando a precisão dos cálculos de força atômica com a transformação de coordenadas de dobra espacial