Um modelo treinado para prever perfis espectroscópicos ajuda a decifrar a estrutura dos materiais
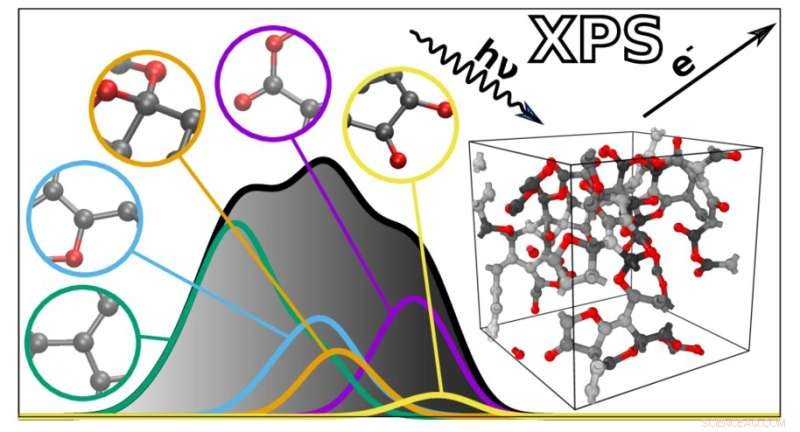
O novo algoritmo prevê os espectros XPS de materiais complexos com base em contribuições atômicas individuais. Crédito:Miguel Caro / Universidade de Aalto
Os materiais à base de carbono têm um enorme potencial para construir um futuro sustentável, mas os cientistas de materiais precisam de ferramentas para analisar adequadamente sua estrutura atômica, que determina suas propriedades funcionais. A espectroscopia de fotoelétrons de raios X (XPS) é uma das ferramentas usadas para fazer isso, mas os resultados do XPS podem ser difíceis de interpretar. Agora, pesquisadores da Aalto desenvolveram uma ferramenta de aprendizado de máquina para melhorar as análises XPS, que eles disponibilizaram gratuitamente como XPS Prediction Server.
Os espectros XPS são gráficos com uma coleção de picos que refletem a energia de ligação dos elétrons nas profundezas dos átomos que compõem um material. Como as energias de ligação dependem do ambiente atômico, elas podem ser usadas para inferir como os átomos estão conectados em um determinado material ou molécula. No entanto, isso também torna os espectros XPS difíceis de interpretar, uma vez que muitos fatores afetam as energias de ligação. As energias de ligação de diferentes características atômicas também podem se sobrepor, complicando ainda mais a análise.
Para ajudar nisso, uma equipe liderada por Miguel Caro desenvolveu um método computacional que pode prever o espectro de energia de ligação de um material com base em um modelo estrutural gerado por computador. Isso simplifica a interpretação dos dados XPS, tornando possível combinar as energias de ligação observadas experimentalmente com as previsões computacionais.
A ideia em si não é nova, mas o problema tem sido a dificuldade computacional de calcular com precisão o espectro XPS de um material. A equipe de Caro resolveu isso usando aprendizado de máquina. O truque era treinar um algoritmo de computador barato para prever o resultado de um método de referência computacionalmente caro com base em uma combinação eficiente de dados mecânicos quânticos computacionalmente baratos e caros.
O método computacionalmente mais barato, DFT, não corresponde aos resultados experimentais com muita precisão. O método mais preciso, GW, leva muito tempo para calcular quando uma molécula tem muitos átomos. "Decidimos construir um modelo de linha de base que usa dados DFT abundantes e depois refiná-lo com dados GW escassos e preciosos. E funcionou", diz Caro.
O algoritmo resultante pode prever o espectro de qualquer material desordenado feito de carbono, hidrogênio e oxigênio. "Os espectros previstos são notavelmente próximos aos obtidos experimentalmente. Isso abre as portas para uma melhor integração entre a caracterização experimental e computacional de materiais", diz Caro. Em seguida, a equipe planeja estender sua técnica para incluir uma gama mais ampla de materiais e outros tipos de espectroscopia.
O artigo de acesso aberto foi publicado em
Chemistry of Materials .
+ Explorar mais O carbono tipo diamante é formado de forma diferente do que se acreditava – o aprendizado de máquina permite o desenvolvimento de um novo modelo