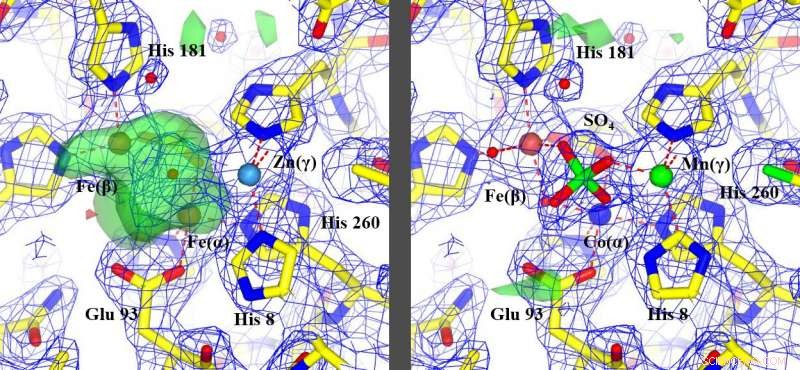
O novo método, corrigindo os metais identificados incorretamente, permitiu a reinterpretação de recursos não identificados, destacado em verde, (imagem à esquerda) para identificar como a proteína funcionava, (imagem à direita). Crédito:Edward Snell
Proteínas que contêm metal, conhecido como metaloproteínas, desempenham papéis importantes na biologia, regulando várias vias no corpo, que muitas vezes se tornam alvos de drogas que salvam vidas. Embora a quantidade de metal nessas proteínas seja geralmente minúscula, é crucial para determinar a função dessas moléculas complexas.
Os cientistas sabem há muito tempo que as metaloproteínas são vitais para a compreensão das doenças, como câncer, e para o desenvolvimento de novos medicamentos, uma vez que os inibidores de metaloproteínas têm sido usados para tratar doenças desde câncer e HIV / AIDS a infecções bacterianas e hipertensão. Mas não houve um confiável, método analítico para determinar a identidade e quantidade de átomos de metal em metaloproteínas.
Agora, em um estudo publicado no mês passado no Jornal da American Chemical Society , uma equipe internacional de pesquisadores relatou ter desenvolvido uma maneira de identificar e contar de forma inequívoca os átomos de metal em proteínas de uma forma eficiente e rotineira. Usando isso, a equipe, que incluía cientistas da Universidade de Buffalo, Hauptman-Woodward Medical Research Institute e outros - revelaram novas informações que estavam lá, mas anteriormente oculto.
Chamada Partícula Induzida Emissão de Raios-X, ou PIXE, o método foi desenvolvido pela primeira vez na década de 1990 por Elspeth F. Garman da University of Oxford e Geoffrey W. Grime do University of Surrey Ion Beam Center, ambos os autores no artigo atual.
O avanço relatado neste artigo é o desenvolvimento do método em uma abordagem eficiente de alto rendimento e a combinação com outros dados experimentais para identificar o tipo e a posição precisa dos metais nas proteínas. Isso permite muitos tipos diferentes de proteínas, grande número deles compõe a vida como a conhecemos, para ser analisado de forma rápida e eficiente, e fornece novas informações para um melhor entendimento estrutural.
A equipe aplicou o novo método a 30 metaloproteínas selecionadas aleatoriamente, que já estão no repositório global de estruturas de proteínas, denominado Protein Data Bank. O que aconteceu a seguir os chocou.
“O resultado foi impressionante '
"Eu sentei em Buffalo com meu colaborador de Oxford e quando analisamos os números, nós dois imediatamente percebemos que tínhamos feito uma descoberta, "lembrou Edward Snell, Ph.D., um dos autores correspondentes, que é presidente e CEO da Hauptman-Woodward e professor do Departamento de Design e Inovação de Materiais, um programa conjunto da Escola de Engenharia e Ciências Aplicadas da UB e sua Faculdade de Artes e Ciências. "Transformamos os números em uma imagem e, escondida nos dados, havia uma explicação de como essa máquina molecular funcionava.
"Fomos os primeiros no mundo a ver o que estava escondido ali o tempo todo. O resultado foi impressionante."
Os resultados mostraram que os métodos usados anteriormente para determinar algumas dessas 30 estruturas de proteínas aleatórias identificaram erroneamente o átomo de metal ou, em alguns casos, perdi completamente.
"De acordo com nossos resultados, o conhecimento atual de cerca de metade das amostras que estudamos está incorreto, "disse Snell.
Os pesquisadores observaram que o Protein Data Bank é um recurso crítico para pesquisadores de todo o mundo. Em 2017, houve em média 1,86 milhão de downloads por dia somente nos EUA. Eles observam que um grande número de pesquisadores usa estruturas do banco de dados sem conhecimento dos possíveis erros fundamentais que podem estar presentes.
E atualmente, mais de 30% dos modelos de banco de dados contêm um metal.
Implicações profundas
"Extrapolando nossos resultados em que havia um metal identificado incorretamente em pelo menos metade das amostras estudadas sugere que mais de 350, 000 modelos baixados por dia podem não conter o metal correto, "Snell disse." Isso tem implicações profundas para aqueles que usam os modelos. Se esses modelos estiverem errados, a compreensão de milhões de pessoas que os usam torna-se falha. "
Snell explicou que uma das dificuldades em estudar metais em proteínas é que eles são muito sensíveis à radiação de raios-X, portanto, o próprio experimento pode mudar o que você vê. Mas ele notou, uma técnica que usa lasers de elétrons livres de raios-X (XFELs), evita isso porque os experimentos são geralmente mais rápidos do que qualquer mudança que possa ocorrer.
Snell dirige o Centro de Ciência e Tecnologia BioXFEL da National Science Foundation, (Biologia com lasers de elétrons livres de raios-X), um consórcio da UB, Hauptman-Woodward e seus parceiros. O centro é dedicado ao uso de XFELs, que produzem raios-X incrivelmente intensos em pulsos extremamente curtos, e pode ajudar no entendimento preciso desses metais em sistemas biológicos.
Com base em sua experiência com o centro de triagem de cristalização de alto rendimento da Hauptman-Woodward, Snell colaborou para implementar a técnica PIXE em um ambiente de alto rendimento. Ele usou seu conhecimento das propriedades dos raios X para identificar que novas informações estruturais estavam presentes nos dados e então pegou esse conhecimento e o transformou em um resultado estrutural.
"Basicamente, meus colegas identificaram os metais e nosso trabalho em Buffalo mostrou a eles onde colocá-los, revelando as novas informações que se tornaram disponíveis quando o metal no modelo estava correto, " ele disse.