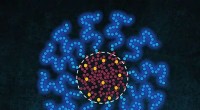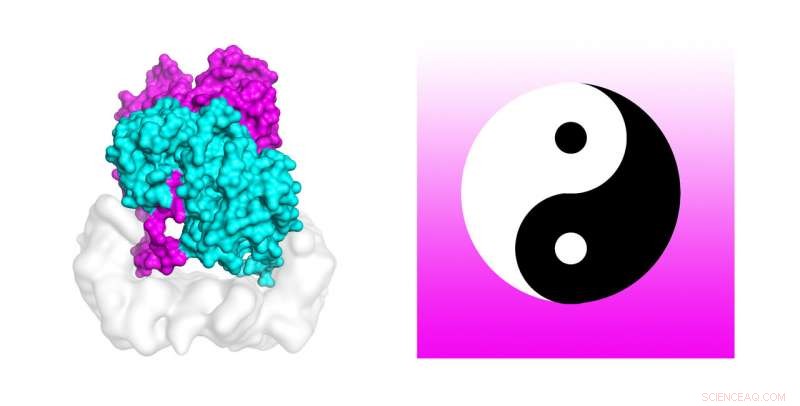
Os cientistas estão usando supercomputadores poderosos para descobrir o mecanismo que ativa as mutações celulares encontradas em cerca de 50 por cento dos melanomas. Simulações de dinâmica molecular no supercomputador Stampede2 da TACC testaram a estabilidade da estrutura do complexo B-Raf:14-3-3, que, quando sofre mutação, está ligada ao câncer de pele. Os autores do estudo comparam o dímero B-Raf ao símbolo circular chinês yin-yang de opostos interconectados unidos pela cauda. Crédito:Karandur et al., TACC
Começa pequeno, apenas uma mancha na pele. As manchas mais comuns permanecem assim - aglomerados inofensivos de células da pele chamados melanócitos, que nos dão pigmento. Em casos raros, o que começa como uma toupeira pode se transformar em melanoma, o tipo mais grave de câncer de pele humano porque pode se espalhar por todo o corpo.
Os cientistas estão usando supercomputadores poderosos para descobrir o mecanismo que ativa as mutações celulares encontradas em cerca de 50 por cento dos melanomas. Os cientistas dizem estar esperançosos de que seu estudo possa ajudar a levar a uma melhor compreensão do câncer de pele e ao desenvolvimento de medicamentos melhores.
Em 2002, os cientistas encontraram uma ligação entre o câncer de pele e as mutações da quinase B-Raf (Fibrossarcoma Rapidly Accelerated), uma proteína que faz parte da cadeia de sinal que começa fora da célula e vai para dentro para direcionar o crescimento celular. Este caminho de sinal, chamada de via da quinase Ras / Raf / Mek / Erk, é importante para a pesquisa do câncer, que visa compreender o crescimento celular fora de controle. De acordo com o estudo, cerca de 50 por cento dos melanomas têm uma única mutação específica no B-Raf, conhecido como resíduo de valina 600 em glutamato (V600E).
O B-Raf V600E, portanto, tornou-se um importante alvo de drogas, e inibidores específicos do mutante foram desenvolvidos nos anos seguintes. As drogas inibiram o mutante, mas algo estranho aconteceu. Paradoxalmente, acalmar o mutante tinha um lado negativo. Ele ativou o não-mutado, proteína quinases B-Raf de tipo selvagem, que novamente desencadeou o melanoma.
"Com este pano de fundo, trabalhamos no estudo da estrutura desta importante proteína, B-Raf, "disse Yasushi Kondo, um pesquisador de pós-doutorado no John Kuriyan Lab na UC Berkeley. Kondo é coautor de um estudo de outubro de 2019 publicado na revista Ciência que determinou a estrutura do complexo de proteínas que compõem o B-Raf e também descobriu como acontece a paradoxal ativação do B-Raf.
"Nosso objetivo foi estudar o estado mais parecido com o nativo da proteína para entender como ela é regulada nas células, porque a maioria dos estudos tem se concentrado no domínio isolado da quinase e como as drogas se ligam ao domínio da quinase ", disse Kondo.
A proteína B-Raf de comprimento total é feita de vários domínios ligados por regiões desordenadas, algo muito pesado para os cientistas ainda imaginarem. A técnica de Kondo era usar química intein para fazer fragmentos menores, em seguida, costure-os para obter a estrutura completa.
"Como resultado, obtivemos uma forma ativa do dímero B-Raf de comprimento total denominado B-Raf co-purificado com dímero 14-3-3, uma proteína de andaime ligada à cauda B-Raf C-terminal fosforilada, "Kondo disse.
O grupo de Kondo usou microscopia crioeletrônica (crio-EM) para determinar a estrutura do complexo B-Raf 14-3-3, basicamente congelando criogenicamente o complexo de proteínas, que o manteve em um quimicamente ativo, ambiente quase natural. Em seguida, eles o projetaram com feixes de elétrons para obter milhares de 'quadros congelados'. Eles peneiraram o ruído de fundo e reconstruíram mapas de densidade tridimensionais que mostravam detalhes até então desconhecidos na forma da molécula. E para proteínas, a forma segue a função.
Kondo explicou que a estrutura revelava uma organização assimétrica do complexo, formado por dois conjuntos de dímeros internamente simétricos, ou pares de moléculas ligadas. "Propomos que este arranjo inesperado permite a ativação assimétrica do dímero B-Raf, que é um mecanismo que fornece uma explicação da origem da ativação paradoxal de B-Raf por inibidores de pequenas moléculas, "Kondo disse.

O supercomputador Stampede2 no Texas Advanced Computing Center é um recurso alocado do Extreme Science and Engineering Discovery Environment (XSEDE), financiado pela National Science Foundation (NSF). Crédito:TACC
A análise detalhada da estrutura complexa assimétrica do B-Raf 14-3-3 mostrou outra característica estrutural inesperada, descrito como o segmento distal da cauda, DTS para abreviar, de uma molécula B-Raf. Kondo disse que a cauda de um está ligada ao local ativo do outro, bloqueando sua atividade competindo com a ligação de ATP. A molécula B-Raf bloqueada é estabilizada na conformação ativa. "Nós interpretamos esta estrutura que esta molécula B-Raf bloqueada funciona como um ativador e estabiliza o outro receptor B-Raf através da interface do dímero, "Kondo disse.
Suficientemente curioso, os autores comparam o dímero B-Raf ao símbolo circular chinês yin-yang de opostos interconectados unidos na cauda. "Olhando para o assunto, é muito claro que não é possível fosforilar a molécula a jusante, que é necessário para o crescimento celular. A outra molécula é claramente aquela que faz o trabalho. Neste conjunto de duas moléculas, vemos claramente que um está fazendo o trabalho de suporte, e o outro está fazendo o trabalho real. Realmente se parece com Yin e Yang neste complexo B-Raf 14-3-3 que resolvemos, "Kondo disse.
Visual, no entanto, pode ser enganoso. Os cientistas usaram simulações de computador para ajudar a verificar se eles estavam realmente certos. "Fizemos simulações de dinâmica molecular deste complexo do dímero B-Raf ligado a um dímero 14-3-3 para testar a estabilidade da conformação assimétrica, "disse o co-autor do estudo Deepti Karandur, também pesquisador de pós-doutorado no John Kuriyan Lab da UC Berkeley; ela também é pós-doutoranda no Howard Hughes Medical Institute. "Não sabíamos porque a conformação era assimétrica, ou qual o papel que desempenhou na manutenção do estado ativo da enzima, "Karandur disse.
Eles começaram as simulações usando a estrutura que Kondo havia resolvido por crio-EM, com o segmento DTS correndo de uma quinase para o local ativo da outra. Em seguida, eles executaram um segundo conjunto de simulações com o segmento DTS removido.
"O que descobrimos foi que no sistema sem o segmento distal da cauda, todo o complexo não é estável, "Karandur explicou." Os domínios da quinase se movem em relação ao andaime, o dímero 14-3-3. Em uma de nossas simulações, o estado dímero do próprio B-Raf, que experimentos mostraram ser necessário para manter o estado ativo desta quinase, desmoronou, indicando que este segmento distal da cauda, DTS, é necessário para realmente manter este complexo nesta conformação assimétrica, que por sua vez é necessário para manter o dímero de quinase no estado ativo de dímero assimétrico estável. "
Um dos principais resultados do estudo foi encontrar o mecanismo de ação que ativa o complexo de quinase B-Raf de duas quinases B-Raf e duas proteínas de andaime 14-3-3, onde na quinase B-Raf está o ativador, e o outro é o receptor.
"A cauda da molécula receptora está dentro do sítio ativo do ativador, então o ativador não pode funcionar como uma enzima, "Kondo disse." Em vez disso, a molécula ativadora estabiliza a conformação ativa da molécula receptora. A proteína-esqueleto 14-3-3 facilita esse arranjo, de modo que a inserção da cauda só acontece com uma molécula de quinase. Nossa hipótese é que, quando não há ligação 14-3-3, ambas as quinases podem ser bloqueadas pela inserção do DTS, mas isso precisa ser testado. "
Os desafios computacionais do estudo envolveram simulações de dinâmica molecular que modelaram a proteína em nível atômico, determinar as forças de cada átomo em todos os outros átomos para um sistema de cerca de 200, 000 átomos em intervalos de tempo de dois femtossegundos.
"Para sistemas pequenos, podemos ver o que está acontecendo de forma relativamente rápida, mas para grandes sistemas como estes, especialmente grandes sistemas biomoleculares, essas mudanças acontecem em escalas de tempo de nanossegundos, escalas de tempo de microssegundos, ou mesmo escalas de tempo de milissegundos, "Karandur disse.
Karandur e colegas recorreram ao XSEDE, o Extreme Science and Engineering Discovery Environment financiado pela NSF, para alocação de tempo no supercomputador Stampede2 no Texas Advanced Computing Center (TACC) para fazer as simulações, bem como o sistema Bridges no Pittsburgh Supercomputer Center para investigar outras proteínas na via. Nós do processador Skylake do Stampede2, em rede com o Intel Omnipath, fez um trabalho rápido com as simulações de dinâmica molecular NAMD otimizadas para supercomputadores.
"Stampede2 funciona muito, muito rápido, e é muito eficiente. Geramos um total de cerca de 1,5 microssegundo de trajetórias para nossos sistemas em cerca de quatro a seis semanas. Enquanto que, se o executássemos em nosso próprio cluster interno, teria levado meses ou mais, "Karandur disse.
Sobre XSEDE, Karandur comentou:"Acho que é um recurso incrível. Tenho feito simulações desde quando era estudante de graduação. O XSEDE possibilitou que acessássemos escalas de tempo que são biologicamente relevantes. Tudo o que acontece em uma célula, acontece em escalas de tempo de microssegundos, para escalas de tempo de milissegundos, para mais. Quando eu estava começando, não pudemos executar esta simulação em nenhum sistema em qualquer lugar. Quero dizer, levaria cinco anos, ou mais. Ser capaz de fazer isso em semanas e dizer, Certo, sabemos entender por que isso é importante para que possamos agora começar a obter uma compreensão real de como a biologia acontece, é simplesmente incrível, "Karandur disse.
E ainda há muito a ser descoberto sobre o B-Raf. É apenas um elo da cadeia de sinal que governa o crescimento celular e o câncer.
“A estrutura que foi resolvida neste artigo faz parte de um grande, sistema multi-domínio, "Karandur explicou." Não sabemos como é esta proteína completa. Não vemos isso na estrutura. Não sabemos como é sua dinâmica, e como todas essas outras partes da proteína desempenham um papel na manutenção do estado ativo, ou convertendo-o do estado inativo para o estado ativo. "
Ela promoveu que, à medida que o sistema fica maior, as mudanças estruturais pertinentes acontecem em escalas de tempo mais longas, e supercomputadores maiores são necessários para lidar com a complexidade, como o supercomputador Frontera, financiado pela NSF, também na TACC.
"O Frontera está chegando lá. Estamos muito animados com isso. Estamos no processo de obter uma alocação no Frontera, "Karandur disse.
Para não cientistas, essa pesquisa fundamental pode fornecer informações que levem a melhores medicamentos para o câncer de pele.
"A ativação paradoxal da quinase Raf por esses inibidores específicos de B-Raf transformam células normais em tumores durante o tratamento do câncer de pele, "Kondo disse. Compreender o mecanismo desse fenômeno nos permitirá projetar drogas melhores. Esperançosamente, nosso estudo pode contribuir para a compreensão desta etapa. Além disso, encontramos mutações nesta ligação entre o domínio quinase e o elemento de ligação 14-3-3 da molécula B-Raf, que nunca foi mostrado antes. Esta mutação reduz a atividade do B-Raf nas células. Também está indicando que esta parte do domínio da quinase pode ser um ponto-alvo para desenvolver novos tipos de inibidores B-Raf. "
Disse Karandur:"Há muita dinâmica acontecendo na célula. em grande parte por causa do XSEDE, apenas começando a ser capaz de olhar para coisas assim. Daqui para frente, a única maneira de continuarmos a olhar para as coisas é usando muito, supercomputadores muito grandes, porque os cálculos exigem muito poder computacional. É muito empolgante poder ver essas coisas acontecerem e dizer:aqui estão como as coisas mudam no nível atômico; aqui estão essas interações entre esses dois átomos que se formam ou se quebram, e isso se traduz nesta grande mudança em nível global na estrutura geral da proteína, e como ele interage com outras proteínas, ou outras moléculas na célula. Estamos muito entusiasmados com o que acontecerá no futuro. "
O estudo, "A estrutura Cryo-EM de um complexo B-Raf:14-3-3 dimérico revela assimetria nos sítios ativos das quinases B-Raf, "foi publicado em 4 de outubro, 2019 no jornal Ciência .