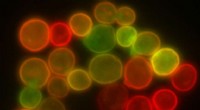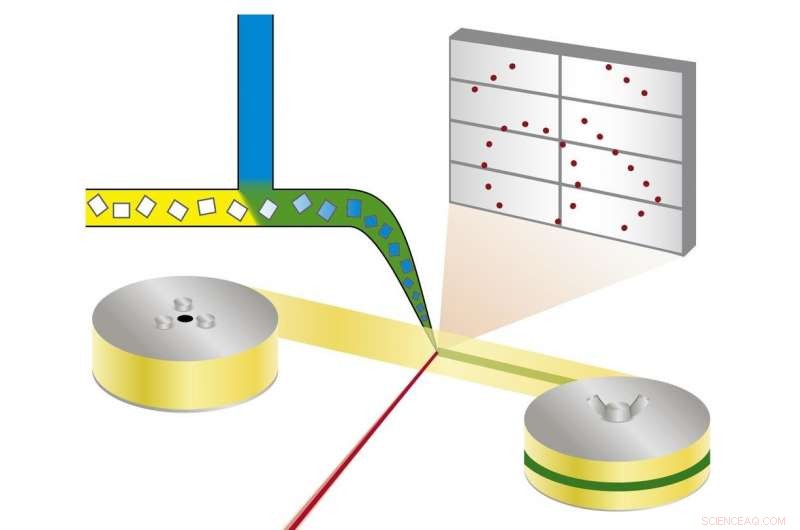
Princípio da cristalografia síncrotron serial mista e difusa:os cristais de proteína são misturados com uma solução de um candidato a medicamento e radiografados em uma fita que passa pelo feixe de raios-X. Crédito:Beyerlein et al., IUCrJ
Os cientistas do DESY desenvolveram um novo método que permite a triagem automatizada e rápida de candidatos a medicamentos promissores. Esta nova técnica, chamada cristalografia síncrotron serial mista e difusa, pode criar imagens da interação de potenciais alvos de drogas com candidatos a drogas ou outras moléculas. O conceito tem o potencial de levar o design de drogas baseadas em estruturas e fragmentos a um novo nível, enquanto os pesquisadores escrevem no Jornal da União Internacional de Cristalografia ( IUCrJ )
Muitas proteínas do corpo são alvos potenciais de drogas. Moléculas farmacêuticas com o formato correto podem se ligar a essas proteínas e ativar ou desativar sua função. Por exemplo, para combater certas formas de leucemia, o medicamento contra o câncer Imatinib inibe uma variante hiperativa da enzima tirosina-quinase, uma proteína responsável por ativar muitas outras proteínas. Imatinib bloqueia o sítio ativo desta tirosina-quinase. Para conseguir isso, a molécula do medicamento deve se encaixar precisamente no local ativo, como uma chave em uma fechadura. Com base no conhecimento da estrutura espacial do alvo da enzima, O imatinibe foi feito sob medida para esse fim.
"Esta estratégia é chamada de design de drogas com base na estrutura e hoje em dia é usada como um método padrão no desenvolvimento de drogas farmacêuticas, "explica o primeiro autor Kenneth Beyerlein do Center for Free-Electron Laser Science (CFEL), uma cooperação de DESY, a Universidade de Hamburgo e a Sociedade Alemã Max Planck. "Contudo, na realidade, alvejar proteínas é muito mais complexo do que colocar uma chave na fechadura. Portanto, muitas moléculas farmacêuticas potenciais ou fragmentos de tais moléculas têm que ser testados, que geralmente é um procedimento demorado e complicado. "Além disso, biólogos e farmacologistas estão interessados no funcionamento preciso dos agentes naturais que se ligam às proteínas, para entender melhor o mecanismo da vida.
O sistema desenvolvido pela equipe em torno de Beyerlein e seu colega do DESY, Dominik Oberthür, também do CFEL, oferece uma nova maneira de atingir esse objetivo:ele mistura proteínas microcristalinas com moléculas específicas chamadas ligantes que podem ser candidatos a drogas ou agentes naturais antes de sondar os cristais com raios-X para revelar a estrutura espacial detalhada do complexo proteína-ligante resultante ou o ausência de tal complexo se um ligante potencial não se liga à proteína.
Para analisar a estrutura espacial de uma proteína, os cientistas costumam usar cristalografia de raios-X. Para esta técnica, um cristal deve ser cultivado a partir da proteína primeiro. Os pesquisadores então tiram instantâneos de raios-X de todos os lados do cristal que precisa ser resfriado a temperaturas ultrabaixas para reduzir os danos da radiação intensa. Os raios X produzem um padrão de difração característico a partir do qual a estrutura interna do cristal e, portanto, a estrutura espacial da proteína pode ser calculada. Para investigar uma proteína com um ligante, um novo cristal deve ser cultivado a partir de uma solução de proteína e ligante ou o cristal deve ser embebido com o ligante. Mesmo com o uso da robótica para automatizar todas as etapas desse processo, a necessidade de montar cristais individuais para cada novo conjunto de dados tornou-se a etapa limitadora da taxa na triagem de grandes bibliotecas de compostos.
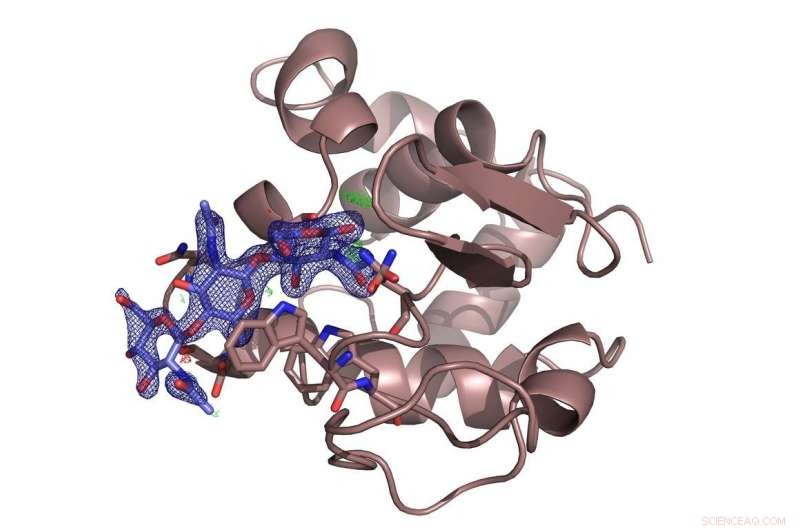
A enzima lisozima (marrom) com o açúcar inibidor quitotriose (azul) ligada a ela. A investigação resolveu uma controvérsia sobre o local de ligação preferido da molécula de açúcar. Crédito:DESY, Dominik Oberthür
A nova técnica segue uma abordagem diferente. "Estamos usando microcristais, que têm duas vantagens:geralmente são muito mais fáceis de produzir do que cristais grandes, e eles são pequenos o suficiente para que uma droga potencial em uma solução possa se difundir através do cristal e se ligar a todas as moléculas de proteína em alguns milissegundos, "explica Oberthür. O sistema desenvolvido pela equipe de Oberthür e Beyerlein dispensa um fluxo de microcristais em um líquido transportador em uma fita fina. Como uma correia transportadora, a fita carrega os cristais através do feixe de raios-X, que é cortado em breves flashes por uma cortina giratória. Em vez de girar um grande cristal no feixe de raios-X, muitos microcristais em orientação aleatória são, portanto, submetidos a raios-X de uma maneira serial e os padrões de difração de cada foto são posteriormente combinados para formar um conjunto de dados completo, seguindo o conceito de cristalografia serial que foi desenvolvido pela primeira vez em lasers de raios-X de elétrons livres (XFELs).
Por meio de uma segunda válvula no sistema, é adicionada uma solução de um candidato a fármaco ou ligando natural. O ponto onde os dois líquidos se misturam pode ser ajustado para criar um atraso definido antes de investigar a estrutura. Esta configuração não requer crio-resfriamento de cristais, portanto, a interação proteína-droga pode ser observada em temperaturas fisiológicas, ou qualquer outra temperatura desejada. Por aqui, mesmo a dinâmica de ligação pode ser investigada. "Podemos difundir produtos químicos nos cristais de proteína instantaneamente e observar a ligação acontecer, "explica Oberthür." Você não precisa encontrar novas condições de crescimento para cada inibidor e não precisa trocar os cristais manualmente, todo o processo pode ser automatizado. "
A equipe testou o novo sistema na fonte de raios-X de alto brilho da DESY, PETRA III, com a conhecida lisozima protéica e uma molécula de açúcar, quitotriose, que inibe a enzima. Os microcristais de lisozima usados aqui tinham apenas cerca de seis a oito micrômetros de diâmetro. A configuração na estação de medição P11 revelou a estrutura espacial do inibidor misturado ligado à lisozima em detalhes. E embora a estrutura da lisozima tenha sido a primeira estrutura enzimática revelada pela cristalografia de raios-X há 50 anos, o novo método ainda pode revelar novos detalhes sobre o modo de ligação da quitotriose à lisozima, resolver uma controvérsia sobre o local de ligação preferido da molécula de açúcar.
Embora a prova de princípio ainda levasse algum tempo, a rotina e os avanços no detector e na tecnologia de raios-X irão acelerar consideravelmente o procedimento. Também, usando todo o espectro do feixe de raios-X da fonte de luz síncrotron em vez de apenas uma única "cor" dela, pode empurrar o tempo de exposição para imagens de difração individuais até 100 picossegundos, ou 0,1 bilionésimos de segundo. Apenas 50 dessas imagens são suficientes para determinar a estrutura, como foi mostrado recentemente.
"Estamos desenvolvendo maneiras de resolver a estrutura das proteínas ligadas para a descoberta de medicamentos de alto rendimento, "explicou Beyerlein. Como as fontes de luz síncrotron são mais acessíveis do que os lasers de raios-X, os pesquisadores imaginam usar este método para triagem rotineira através de bibliotecas de inibidores potenciais e fragmentos de drogas. "Fazer isso automaticamente e muito mais rápido do que com as abordagens convencionais seria um grande passo à frente no design de medicamentos com base na estrutura, "diz Beyerlein.