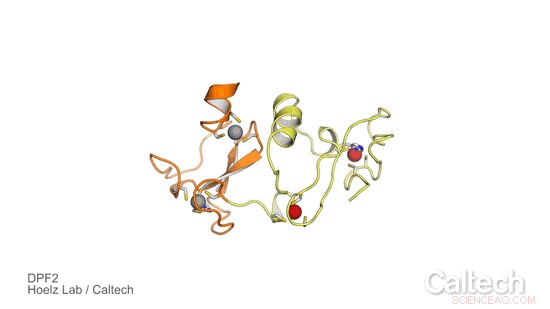
Uma estrutura de cristal de uma porção do DPF2 humano, uma proteína que controla um interruptor genético que diz às células-tronco do sangue quando se tornarem glóbulos vermelhos e brancos. As regiões laranja e amarela ilustram o domínio DPF2 'leitor', que é estabilizado por íons de zinco, representado como esferas vermelhas e cinzas. Crédito:Hoelz Lab / Caltech
Muitas coisas dão errado nas células durante o desenvolvimento do câncer. No centro do caos, muitas vezes estão os interruptores genéticos que controlam a produção de novas células. Em uma forma particularmente agressiva de leucemia, chamada leucemia mieloide aguda, uma mudança genética que regula a maturação das células-tronco do sangue em células vermelhas e brancas do sangue dá errado. Normalmente, essa mudança leva a um número apropriado de glóbulos brancos e vermelhos. Mas os pacientes com leucemia mieloide aguda acabam com um perigoso acúmulo de células-tronco do sangue e uma falta de células vermelhas e brancas do sangue - células que são necessárias para fornecer oxigênio ao corpo e combater infecções.
Agora, pesquisadores da Caltech e do Sylvester Comprehensive Cancer Center da Universidade de Miami estão se concentrando em uma proteína que ajuda a controlar essa mudança genética. Em indivíduos saudáveis, a proteína, chamado DPF2, interrompe a produção de glóbulos vermelhos e brancos quando não precisam ser substituídos. Isso é, ele desliga o interruptor. Mas a proteína pode ser superproduzida em pacientes com leucemia mieloide aguda. A proteína basicamente fica no interruptor, impedindo-o de voltar a formar as células sanguíneas conforme necessário. Pacientes com superprodução de DPF2 têm um prognóstico particularmente ruim.
Em um novo estudo, a ser publicado na semana de 22 de maio, 2017, no jornal Proceedings of the National Academy of Sciences , os pesquisadores demonstram novas maneiras de impedir o DPF2, potencialmente tornando a leucemia mieloide aguda mais tratável. Eles relatam novos detalhes estruturais e funcionais sobre um fragmento de DPF2. Essas novas informações revelam alvos para o desenvolvimento de drogas que bloqueariam a função da proteína.
"Muitas doenças humanas, incluindo câncer, surgem por causa de interruptores genéticos com defeito, "diz André Hoelz, o autor correspondente do estudo. Hoelz é professor de química na Caltech, um investigador do Heritage Medical Research Institute (HMRI), e um bolsista do corpo docente do Howard Hughes Medical Institute (HHMI). "Elucidar como eles funcionam nos detalhes atômicos nos permite iniciar o processo de customização de medicamentos para desativá-los e, em muitos casos, isso é um passo significativo para a cura."
Os glóbulos vermelhos e brancos são constantemente regenerados a partir de células-tronco do sangue, que residem em nossa medula óssea. Como outras células-tronco, as células-tronco do sangue podem viver para sempre. É apenas quando eles se diferenciam em tipos específicos de células, como glóbulos vermelhos e brancos, que eles então se tornam mortais, ou adquirir a capacidade de morrer após um certo período de tempo.
"Nossos corpos usam uma série complexa de interruptores genéticos para diferenciar uma célula-tronco do sangue em muitos tipos diferentes de células. Essas células diferenciadas então circulam no sangue e desempenham uma variedade de funções diferentes. Quando essas células chegam ao final de sua vida, elas precisam ser substituído, "diz Hoelz." Isso é um pouco como substituir pneus usados em um carro. "
Para investigar o papel do DPF2 e aprender mais sobre como ele controla a mudança genética para a produção de células sanguíneas, o grupo Hoelz fez parceria com Stephen D. Nimer, co-autor do artigo e diretor do Sylvester Comprehensive Cancer Center, e sua equipe. Primeiro, Ferdinand Huber e Andrew Davenport - ambos estudantes de graduação da Caltech no grupo Hoelz e coautores do novo estudo - obtiveram cristais de uma porção da proteína DPF2 contendo um domínio conhecido como dedo PHD, que significa homeodomínio do planeta. Eles então usaram cristalografia de raios-X, um processo que envolve a exposição de cristais de proteína a raios-X de alta energia, para resolver a estrutura do domínio de dedo PHD. A técnica foi realizada no Stanford Synchrotron Radiation Lightsource, usando uma linha de luz dedicada do Observatório Molecular da Caltech.
Os resultados revelaram como o DPF2 se liga a um complexo DNA-proteína, chamado de nucleossomo, para bloquear a produção de glóbulos vermelhos e brancos. A proteína "lê" vários sinais exibidos na superfície do nucleossomo, adotando uma forma que se ajusta a várias modificações no complexo do nucleossomo, como as peças de diferentes formatos de um quebra-cabeça. Uma vez que a proteína se liga a este locus de DNA, O DPF2 desativa a chave que regula a diferenciação das células sanguíneas.
O próximo passo era ver se o DPF2 poderia ser bloqueado em células-tronco do sangue humano no laboratório. Sarah Greenblatt, um associado de pós-doutorado no grupo de Nimer e co-primeiro autor do estudo, usou a informação estrutural do grupo de Hoelz para criar uma versão mutante da proteína. O grupo Nimer então introduziu a proteína mutada nas células-tronco do sangue, e descobriram que o DPF2 mutado não podia mais se ligar ao nucleossomo. Em outras palavras, O DPF2 não conseguia mais desativar a chave para a produção de células sanguíneas.
"O DPF2 mutado não foi capaz de se ligar a regiões específicas do genoma e não conseguiu interromper a diferenciação de células-tronco do sangue, "diz Huber." Se o DPF2 também pode ser bloqueado nos próprios pacientes com câncer, resta saber. "Os pesquisadores afirmam que existe um encaixe estrutural no DPF2, uma das regiões semelhantes a peças de quebra-cabeça identificadas no novo estudo, é um bom alvo para drogas candidatas.