Adsorção de desdobramento em nanopartículas de metal:conectando estabilidade com catálise
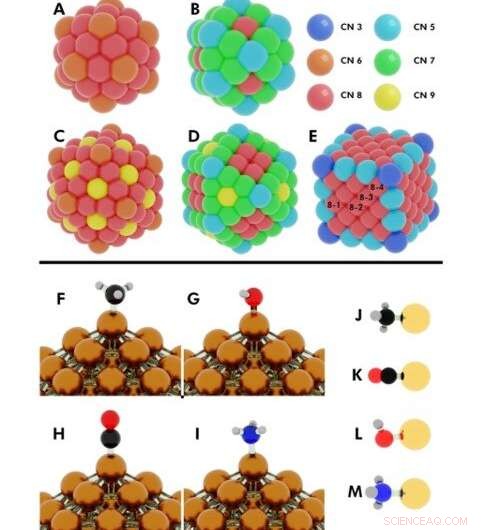 p Ilustração das configurações iniciais para vários cálculos DFT (teoria funcional da densidade) realizados. Superior:números de coordenação em (A) icosaedro de 55 átomos, (B) cuboctaedro de 55 átomos, (C) icosaedro de 147 átomos, (D) cuboctaedro de 147 átomos, (E) cubo de 172 átomos. Nanopartículas (NPs) onde mais de um átomo único compartilham o mesmo número de coordenação (CN), são denotados com números 8-1, 8-2, 8-3, 8-4. Crédito:Science Advances, doi:10.1126 / sciadv.aax5101
p Ilustração das configurações iniciais para vários cálculos DFT (teoria funcional da densidade) realizados. Superior:números de coordenação em (A) icosaedro de 55 átomos, (B) cuboctaedro de 55 átomos, (C) icosaedro de 147 átomos, (D) cuboctaedro de 147 átomos, (E) cubo de 172 átomos. Nanopartículas (NPs) onde mais de um átomo único compartilham o mesmo número de coordenação (CN), são denotados com números 8-1, 8-2, 8-3, 8-4. Crédito:Science Advances, doi:10.1126 / sciadv.aax5101
p Nanopartículas de metal têm recebido atenção substancial devido às suas aplicações em diversos campos da medicina, catálise, energia e meio ambiente. Contudo, as propriedades fundamentais da adsorção de nanopartículas em uma superfície ainda precisam ser compreendidas. James Dean e uma equipe de pesquisa interdisciplinar no departamento de Engenharia Química, nos EUA introduziu um modelo de adsorção universal para levar em conta as características estruturais, composição de metais e diferentes adsorbatos de nanopartículas via aprendizado de máquina (ML). O modelo ajustou um grande número de dados para prever com precisão as tendências de adsorção em nanopartículas monometálicas e baseadas em ligas. O modelo era simples e fornecia dados calculados rapidamente para metais e adsorvatos. A equipe de pesquisa conectou a adsorção com comportamento de estabilidade para avançar no design de nanopartículas ideais para aplicações de interesse. A pesquisa agora está publicada em
Avanços da Ciência . p Nanopartículas de metal (NPs) têm aplicações significativas em catálise, variando de combustível e produção de produtos químicos, à energia solar e química. Mas sua estabilidade e atividade catalítica geralmente mostram tendências opostas, onde catalisadores muito ativos podem operar apenas por alguns ciclos. Uma característica chave na extensão da funcionalidade catalítica metálica depende da força de adsorção para uma variedade de espécies na superfície do catalisador. De acordo com o princípio Sabatier, desenvolvido há mais de um século, os catalisadores ativos devem ligar os adsorvatos com uma força de ligação que não é forte nem fraca. Embora espécies fortemente adsorvidas possam envenenar a superfície do catalisador, reagentes fracamente ligados são facilmente absorvidos. Em um cenário intermediário, os reagentes podem se encontrar e reagir nas superfícies catalíticas. Os pesquisadores atualmente usam simulação computacional e métodos de química teórica para estimular o comportamento catalítico em catalisadores de metal com grande precisão para orientar os experimentos subsequentes em laboratório.
p Os esforços computacionais têm se concentrado na triagem de diferentes catalisadores de metal para descobrir a energia de ligação "mágica" (BE) de espécies químicas em superfícies de catalisador para formar catalisadores muito ativos. O design in silico de materiais cataliticamente ativos, Contudo, continua a ser realizado. As desvantagens são principalmente devido aos esforços de design que muitas vezes negligenciam a estabilidade dos catalisadores. Os catalisadores NP também possuem um alto grau de heterogeneidade de locais em sua superfície para adsorção e catálise. Os cientistas desenvolveram modelos de adsorção para relacionar a energia de ligação dos adsorbatos com as características da superfície dos NPs, como números de coordenação (CNs), para entender a resposta de adsorção específica do local. Ainda, para maior clareza, a variação da energia de ligação (BE) também envolve descritores secundários, como curvatura e propriedades eletrônicas de NPs.
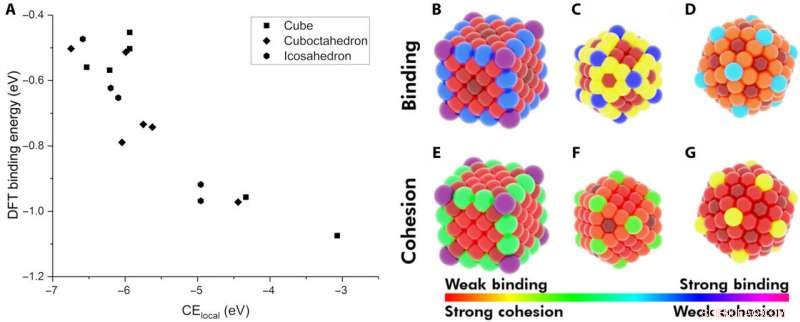 p Demonstração de energia coesiva local (CElocal) como descritor de energia de adsorção. (A) O BE de CO em vários locais de Au NPs como uma função de CElocal:cubo de 172 átomos (retângulos), Icosaedro de 147 átomos (hexágonos), e cuboctaedro de 147 átomos (losango). Mapa de calor de diferentes locais nas NPs com relação ao seu BE de CO (B a D) e ao seu CElocal (E a G). O esquema de cores segue a faixa de ligação do CO mais forte ao CElocal mais fraco (violeta) e da ligação mais fraca ao CElocal mais forte (vermelho). Crédito:Science Advances, doi:10.1126 / sciadv.aax5101
p Demonstração de energia coesiva local (CElocal) como descritor de energia de adsorção. (A) O BE de CO em vários locais de Au NPs como uma função de CElocal:cubo de 172 átomos (retângulos), Icosaedro de 147 átomos (hexágonos), e cuboctaedro de 147 átomos (losango). Mapa de calor de diferentes locais nas NPs com relação ao seu BE de CO (B a D) e ao seu CElocal (E a G). O esquema de cores segue a faixa de ligação do CO mais forte ao CElocal mais fraco (violeta) e da ligação mais fraca ao CElocal mais forte (vermelho). Crédito:Science Advances, doi:10.1126 / sciadv.aax5101
p No presente trabalho, Dean et al. aplicou a teoria funcional da densidade (DFT) e técnicas de aprendizado de máquina para derivar um modelo simples baseado na física para capturar com precisão a energia de adsorção variável. Eles estimaram a variável em função do ambiente do local de adsorção local na superfície do NP, bem como do tipo de metal NP. O modelo generalizado pode ser aplicado a qualquer nanoestrutura de metal para entender o comportamento de adsorção no catalisador NP e a estabilidade do catalisador; para selecionar e projetar catalisadores para inúmeras aplicações.
p Os pesquisadores primeiro hipotetizaram os fatores mais importantes entre NPs monometálicos e adsorbatos. Eles então definiram a energia coesiva local (CE
local ) em metais a granel e CEs capturados em NPs usando um modelo centrado em ligações, que somava todas as energias de ligação metal-metal. Ao aplicar conceitos semelhantes, eles descreveram a estabilidade dos locais de ligação e mostraram como os locais quimicamente insaturados (menos ligações metal-metal) se ligavam aos adsorbatos com uma força aumentada. A equipe de pesquisa se concentrou em descrever a capacidade de ligação de um único par adsorbato-metal. They plotted the DFT-calculated binding energy of carbon monoxide (CO) to a 172-atom gold (Au) cube and a 147-atom gold (Au) cuboctahedron or icosahedron. The team observed a strongly inverse relationship between the local cohesive energy (CE
local ) and binding energy (BE) to suggest the strongest adsorption sites to be those exhibiting the weakest local cohesion.
p The team further developed their model and performed ordinary least squares (OLS) regression to understand adsorption on monometallic NPs and slabs using three adsorbates [Methyl radical (CH
3 ), CO, hydroxyl radical (OH)] on three different metals (Cu, Ag—silver, Au). The metallic NPs contained different morphologies (172-atom cube, 55- and 147-atom icosahedron and 55- and 147-atom cuboctahedron). They observed that the binding affinity to the adsorbates decreased as the cohesion of the local sites increased. And as the adsorbate's chemical potential increased, they became less stable and bound a metal NP with higher tendency. The direct correlation with the metal Adsorbate (MAD) intuitively described the tendency of the metal to bind the adsorbate.
 p Parity plot of the model-predicted binding energy (BE) of adsorbates (OH, CO, and CH3) on various metal systems versus the DFT BE (eV). (A) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which includes the nanoparticle cohesive energy (CENP) term. (B) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which does not include the CENP term. (C) The model trained on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms) and tested against RPBE (revised Perdew-Burke-Ernzerhof model) DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu). (D) The model both trained and tested on RPBE DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu) from the slab dataset. Crédito:Science Advances, doi:10.1126/sciadv.aax5101
p Parity plot of the model-predicted binding energy (BE) of adsorbates (OH, CO, and CH3) on various metal systems versus the DFT BE (eV). (A) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which includes the nanoparticle cohesive energy (CENP) term. (B) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which does not include the CENP term. (C) The model trained on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms) and tested against RPBE (revised Perdew-Burke-Ernzerhof model) DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu). (D) The model both trained and tested on RPBE DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu) from the slab dataset. Crédito:Science Advances, doi:10.1126/sciadv.aax5101
p Dean et al. tested the generalizability of the model and trained the simulation on a single metal or single morphology, although it accurately captured other metals or morphologies as well. The work provided strong evidence that the model captured the underlying physics of the binding interactions, allowing the team to extend the work from non-periodic NPs to periodic slab systems. Computationally inexpensive systems could parameterize the model to extend to larger systems, which was not thus far possible due to the computational costs involved.
p Dean et al. then extended the model from monometallic NPs to bimetallic systems. Para esses experimentos, they plotted the BE—trained on monometallic NPs, across several sites of bi-metallic, 55-atom icosahedron NPs (Cu
31 Ag
24 and Cu
22 Ag
33 ) The model very accurately captured trends in adsorption on the bimetallic Cu/Ag NPs as well. This was an interesting result since the scientists had only trained the model on monometallic systems. The results showed the generalizability of the model for both monometallic and bimetallic NPs. Contudo, the team will account additional descriptors including binding site electronegativity to understand the adsorption behavior for bimetallic systems in depth.
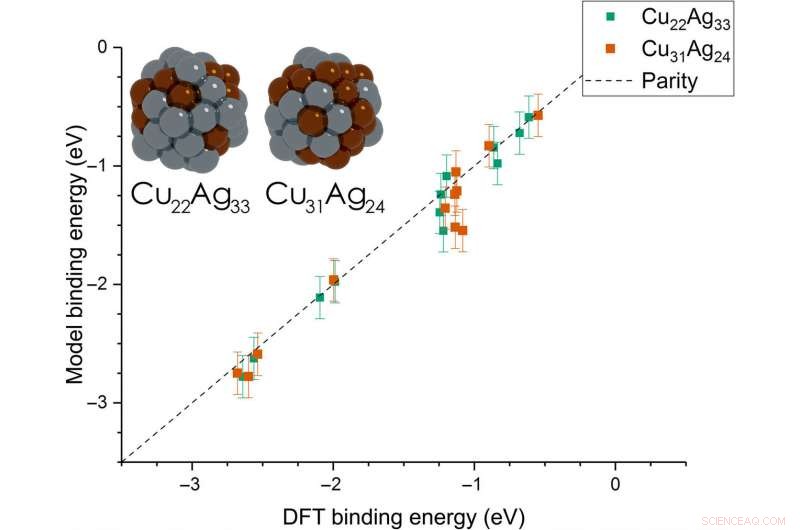 p Parity plot between the presently developed model and DFT calculations on icosahedral bimetallic (Cu55−xAgx, x =24, 33) NPs. The model is trained on CH3, CO, and OH adsorbing on monometallic Ag, Cu, and Au NPs and is able to capture adsorption on bimetallic NPs. Images of the two NPs are shown as inset, with copper and silver atoms colored in brown and gray, respectivamente. Crédito:Science Advances, doi:10.1126/sciadv.aax5101
p Parity plot between the presently developed model and DFT calculations on icosahedral bimetallic (Cu55−xAgx, x =24, 33) NPs. The model is trained on CH3, CO, and OH adsorbing on monometallic Ag, Cu, and Au NPs and is able to capture adsorption on bimetallic NPs. Images of the two NPs are shown as inset, with copper and silver atoms colored in brown and gray, respectivamente. Crédito:Science Advances, doi:10.1126/sciadv.aax5101
p Although Dean et al. trained the ML (machine learning) algorithm to capture the adsorption trends of just one type of d9 metal, it could accurately predict the behavior of similar d9 metals (Cu—coper, Ag and Au). When they trained the model on a dataset of CH
3, CO and OH adsorbed to Cu, Ag and Au NPs, they could also capture general adsorption trends for similar elements in other columns of the periodic table. They then improved the complexity of the machine learning techniques to provide additional avenues to improve the model of adsorption.
p Desta maneira, James Dean and his colleagues developed a simple yet powerful physics-based model to capture trends on the strength of binding interactions between different adsorbates and metal NPs using machine learning techniques. The study was the first to develop an adsorption model that accurately connected the properties of diverse metal NPs with the stability of the adsorption site. The model introduced simple descriptors to capture the adsorption on any site, relative to monometallic and bimetallic NPs. The team generalized the model to effectively stimulate a range of binding interactions, including variations on the types of metals, their composition, sites of adsorption and adsorbates.
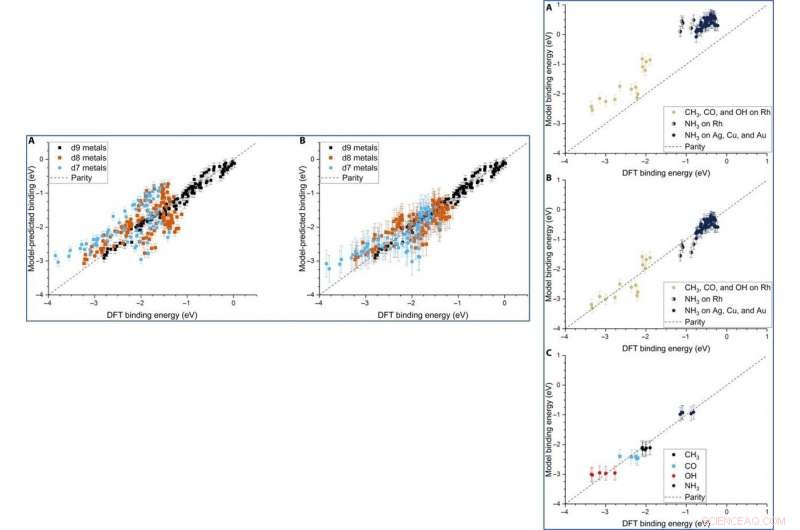 p LEFT:The three-descriptor model extended to slab dataset. (A) The model trained on the slab dataset on Cu, Ag, and Au surfaces and tested against the Rh, Ir, Ni, Pd, Pt, Cu, Ag, and Au surfaces from the slab dataset. (B) The equivalent model when trained separately for each column of the d-block, still using the slab dataset. Error bars in every case are the 10-fold cross-validated RMSE of the training set. RIGHT:Extension of the model to Rh and NH3. (A) The model parameterized on our Ag, Cu, and Au NPs adsorbing CH3, CO, and OH and tested against Rh and NH3. (B) The equivalent model with empirical (constant) corrections for Rh and NH3. In the case of NH3 bound to Rh, both corrections are simultaneously applied and indicated by two-colored dots. (C) The model trained on CH3, CO, OH, and NH3 adsorbing on icosahedral/cuboctahedral Rh55. Crédito:Science Advances, doi:10.1126/sciadv.aax5101
p LEFT:The three-descriptor model extended to slab dataset. (A) The model trained on the slab dataset on Cu, Ag, and Au surfaces and tested against the Rh, Ir, Ni, Pd, Pt, Cu, Ag, and Au surfaces from the slab dataset. (B) The equivalent model when trained separately for each column of the d-block, still using the slab dataset. Error bars in every case are the 10-fold cross-validated RMSE of the training set. RIGHT:Extension of the model to Rh and NH3. (A) The model parameterized on our Ag, Cu, and Au NPs adsorbing CH3, CO, and OH and tested against Rh and NH3. (B) The equivalent model with empirical (constant) corrections for Rh and NH3. In the case of NH3 bound to Rh, both corrections are simultaneously applied and indicated by two-colored dots. (C) The model trained on CH3, CO, OH, and NH3 adsorbing on icosahedral/cuboctahedral Rh55. Crédito:Science Advances, doi:10.1126/sciadv.aax5101
p Although the team did not test the applicability of the model for ternary systems, the physical properties may remain relevant to accurately model multimetallic systems as well. The adsorption model can accurately describe the binding strength of a variety of molecules on any site of NPs, including alloys. The scientists expect the model to be highly applicable as a screening tool for the high throughput search of potential catalysts. p © 2019 Science X Network