Os trios de átomos de tungstênio são muito influenciados em sua migração pela natureza de uma minúscula partícula pela forma da partícula, de acordo com uma equipe de especialistas, incluindo o Dr. Fei Gao do Pacific Northwest National Laboratory. A equipe dos EUA e China realizou simulações computacionais complexas para determinar a energética envolvida na migração do cluster de tungstênio. Eles descobriram que de 3 a 4 adatom, ou átomo de superfície, os aglomerados preferem formar ilhas compactadas. A reorientação é o mecanismo de migração dominante para o dímero, enquanto a migração líquida de clusters de lager pode ser realizada pelo cisalhamento do dímero, movimentos combinados e mecanismos de rotação.
A pesquisa foi destacada na capa do European Physical Journal B em março de 2011, juntamente com o artigo revisado por pares:"Tungsten Clusters Migration on Nanoparticles:A Dimer Method Study."
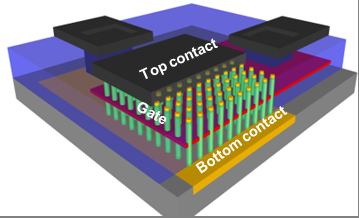
A demanda por miniaturização de dispositivos eletrônicos se beneficiará de uma compreensão mais aprofundada dos materiais nanoestruturados. O tungstênio tem propriedades únicas, como alta densidade, dureza, temperatura de fusão, elasticidade e condutividade, junto com baixa expansão térmica. Essas propriedades únicas e partículas de tamanho nanométrico podem ser usadas para armazenar e organizar elétrons para uso por semicondutores, fornecendo aos engenheiros um material de menor resistência e melhor condutividade.
Usando supercomputadores no Laboratório de Ciências Moleculares Ambientais, a equipe de pesquisa realizou os cálculos necessários para pesquisar possíveis estados de transição e caminhos de migração para aglomerados de tungstênio em nanopartículas de tungstênio, e energias de migração correspondentes para os possíveis caminhos de migração desses aglomerados.
Aglomerados de tungstênio com até quatro adátomos preferem estruturas compactas 2D com energias de ligação relativamente baixas. A equipe determinou que o efeito das regiões de interface e vértice no comportamento de migração dos clusters é significativamente forte em comparação com o tamanho das nanopartículas.
Os mecanismos de migração são muito diferentes quando os clusters estão localizados no centro da nanopartícula e perto da interface ou áreas de vértice. Perto das interfaces e áreas de vértice, os átomos do substrato tendem a participar dos processos de migração dos aglomerados, e pode juntar os adátomos para formar um cluster maior ou levar à dissociação de um cluster por meio do mecanismo de troca, o que resulta no adatom cruzando as facetas.
As barreiras de energia calculadas para os trímeros sugerem que a migração combinada é mais provável do que o salto sucessivo de um único adatom nos aglomerados.
O método computacional multi-escala, variando de cálculo ab initio a método de dinâmica de longo tempo, será ainda empregado para estudar a evolução estrutural de aglomerados de metal de tamanho nanométrico com tamanho crescente e transformação de fase desses aglomerados de metal. Esses estudos fornecerão percepções significativas sobre catalisadores em nanoescala, sensores e aplicações eletrocrômicas, como vidro inteligente, onde as propriedades de transmissão de luz ou calor do vidro são alteradas pela aplicação de voltagem.