Novo algoritmo quântico resolve problema crítico de química quântica por meio da adaptação ao longo de um caminho geométrico
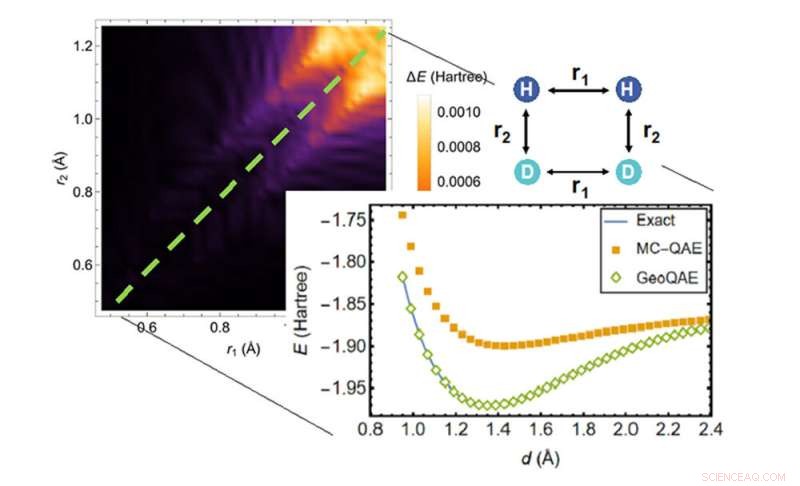
Ao calcular a superfície de energia potencial da reação química de H2;+ D2 → 2HD, o novo algoritmo (losangos verdes) supera o algoritmo anterior (quadrados laranja) na busca da solução mais precisa (linha azul). Crédito:Laboratório Nacional de Brookhaven
Uma equipe de pesquisadores do Laboratório Nacional Brookhaven do Departamento de Energia dos EUA (DOE) e da Universidade Stony Brook desenvolveu um novo algoritmo quântico para calcular as energias mais baixas de moléculas em configurações específicas durante reações químicas, inclusive quando suas ligações químicas são quebradas. Conforme descrito em
Pesquisa de Revisão Física , em comparação com algoritmos existentes semelhantes, incluindo o método anterior da equipe, o novo algoritmo melhorará significativamente a capacidade dos cientistas de calcular com precisão e confiabilidade a superfície de energia potencial nas moléculas em reação.
Para este trabalho, Deyu Lu, físico do Centro de Nanomateriais Funcionais (CFN) do Brookhaven Lab, trabalhou com Tzu-Chieh Wei, professor associado especializado em ciência da informação quântica no C.N. Instituto Yang de Física Teórica da Stony Brook University, Qin Wu, teórico do CFN, e Hongye Yu, Ph.D. estudante em Stony Brook.
“Compreender a mecânica quântica de uma molécula, como ela se comporta em nível atômico, pode fornecer informações importantes sobre suas propriedades químicas, como estabilidade e reatividade”, disse Lu.
Uma propriedade particular que tem sido um desafio para determinar é o estado fundamental de uma molécula:o ponto em que a energia eletrônica total da molécula (incluindo energia cinética e potencial) está no seu nível mais baixo e nada fora desse "sistema molecular" está excitando ou carregando a molécula. elétrons. Quando a estrutura atômica de um sistema químico se torna mais complexa, como em uma grande molécula, muitos mais elétrons podem interagir. Essas interações tornam o cálculo do estado fundamental de moléculas complexas extremamente difícil.
O novo algoritmo quântico melhora o algoritmo anterior para resolver esse problema de maneira criativa. Ele explora uma deformação geométrica suave feita pela variação contínua de comprimentos de ligação ou ângulos de ligação na estrutura da molécula. Com essa abordagem, os cientistas dizem que podem calcular o estado fundamental das moléculas com muita precisão, mesmo quando as ligações químicas estão se quebrando e se reformando durante as reações químicas.
Construindo a base "Ao confiar apenas nos métodos tradicionais de computação, esse problema do estado fundamental contém muitas variáveis para resolver - mesmo nos supercomputadores mais poderosos", disse Lu.
Você pode pensar em um algoritmo como um conjunto de etapas para resolver um problema específico. Os computadores clássicos podem executar algoritmos complexos, mas à medida que se tornam maiores e mais envolvidos, podem se tornar muito difíceis ou demorados para os computadores clássicos resolverem de maneira viável. Os computadores quânticos podem acelerar o processo aproveitando as regras da mecânica quântica.
Na computação clássica, os dados são armazenados em bits que têm um valor de 1 ou 0. Um bit quântico, conhecido como qubit, pode ter um valor além de apenas 0 ou 1, pode até ter um valor de 0 e 1, em um chamada superposição quântica. Em princípio, esses qubits mais "flexíveis" podem armazenar uma quantidade maior de informações do que os bits clássicos. Se os cientistas puderem encontrar maneiras de aproveitar a capacidade de transporte de informações dos qubits, o poder da computação pode se expandir exponencialmente com cada qubit adicional.
Qubits, no entanto, são bastante frágeis. Muitas vezes, eles podem quebrar quando a informação está sendo extraída. Quando um dispositivo quântico interage com o ambiente circundante, pode gerar ruído ou interferência que destrói o estado quântico. Mudanças de temperatura, vibrações, interferência eletromagnética e até defeitos de material também podem fazer com que os qubits percam informações.
Para compensar essas armadilhas, os cientistas desenvolveram uma solução híbrida que aproveita os dois algoritmos de computação clássicos, que são mais estáveis e práticos.
Lu e Wei começaram a pesquisar abordagens híbridas de computação clássica e quântica em 2019. Esta doação anual promove a colaboração entre o Brookhaven National Laboratory e a Stony Brook University, financiando iniciativas de pesquisa conjuntas que se alinham com as missões de ambas as instituições. Com este trabalho inicial, Lu e Wei primeiro se concentraram em resolver o problema do estado fundamental substituindo os algoritmos clássicos mais "caros" - aqueles que eram muito mais complexos e exigiam significativamente mais etapas (e mais tempo de computação) para serem concluídos - por quânticos .
Esticando laços, criando novos caminhos Os pesquisadores observam que os algoritmos quânticos existentes apresentam desvantagens para resolver o problema do estado fundamental, incluindo o que Wei e Yu desenvolveram em 2019. Embora alguns algoritmos populares sejam precisos quando uma molécula está em sua geometria de equilíbrio - seu arranjo natural de átomos em três dimensões - esses algoritmos podem se tornar não confiáveis quando as ligações químicas são quebradas em grandes distâncias atômicas. A formação de ligações e a dissociação desempenham um papel em muitas aplicações, como prever quanta energia é necessária para iniciar uma reação química, então os cientistas precisavam de uma maneira de resolver esse problema à medida que as moléculas reagem. Eles precisavam de novos algoritmos quânticos que pudessem descrever a quebra de ligações.
Para esta nova versão do algoritmo, a equipe trabalhou com o Co-design Center for Quantum Advantage (C2QA), liderado pelo Brookhaven-Lab, que foi formado em 2020. Wei contribui para o impulso do software do centro, especializado em algoritmos quânticos. O novo algoritmo da equipe usa uma abordagem adiabática – que faz mudanças graduais – mas com algumas adaptações que garantem que ele permaneça confiável quando as ligações químicas são quebradas.
"Um processo adiabático funciona adaptando gradualmente as condições de um sistema mecânico quântico", explicou Lu. "De certa forma, você está alcançando uma solução em passos muito pequenos. Você evolui o sistema de um modelo simples e solucionável para o alvo final, normalmente um modelo mais difícil. Além do estado fundamental, no entanto, um sistema multieletrônico tem muitos estados excitados em energias mais altas. Esses estados excitados podem representar um desafio ao usar este método para calcular o estado fundamental."
Wei comparou um algoritmo adiabático a dirigir em uma rodovia, "se você estiver viajando de uma cidade para outra, existem vários caminhos para chegar lá, mas você quer encontrar o mais seguro e eficiente".
No caso da química quântica, a chave é encontrar uma "lacuna de energia" grande o suficiente entre o estado fundamental e os estados excitados, onde não existem estados de elétrons. Com uma lacuna grande o suficiente, os veículos na metáfora da rodovia não "cruzarão as faixas", de modo que seus caminhos possam ser traçados com precisão.
"Uma grande lacuna significa que você pode ir mais rápido, então, de certa forma, você está tentando encontrar uma estrada menos movimentada para dirigir mais rápido sem bater em nada", disse Wei.
"Com esses algoritmos, a entrada do caminho é uma solução simples e bem definida da computação clássica", observou Wei. "Também sabemos onde fica a saída - o estado fundamental da molécula - e estávamos tentando encontrar uma maneira de conectá-la à entrada da maneira mais natural, uma linha reta.
"Fizemos isso em nosso primeiro artigo, mas a linha reta tinha bloqueios causados pelo fechamento da lacuna de energia e cruzamento de caminhos. Agora temos uma solução melhor."
Quando os cientistas testaram o algoritmo, eles demonstraram que, mesmo com mudanças finitas no comprimento da ligação, a versão melhorada ainda funcionava com precisão para o estado fundamental.
"Fomos além da nossa zona de conforto, porque a química não é nosso foco", disse Wei. "Mas foi bom encontrar um aplicativo como esse e fomentar esse tipo de colaboração com o CFN. É importante ter diferentes perspectivas de pesquisa."
Ele observou o esforço acumulado de muitas pessoas. "No grande esquema, acho que estamos fazendo uma pequena contribuição, mas isso pode ser uma base para outros trabalhos nesses campos", disse ele. "Esta pesquisa não é apenas fundamental, mas uma ótima ilustração de como diferentes instituições e instalações podem se unir para alavancar suas áreas de especialização".
+ Explorar mais Rumo a um computador quântico que calcula energia molecular