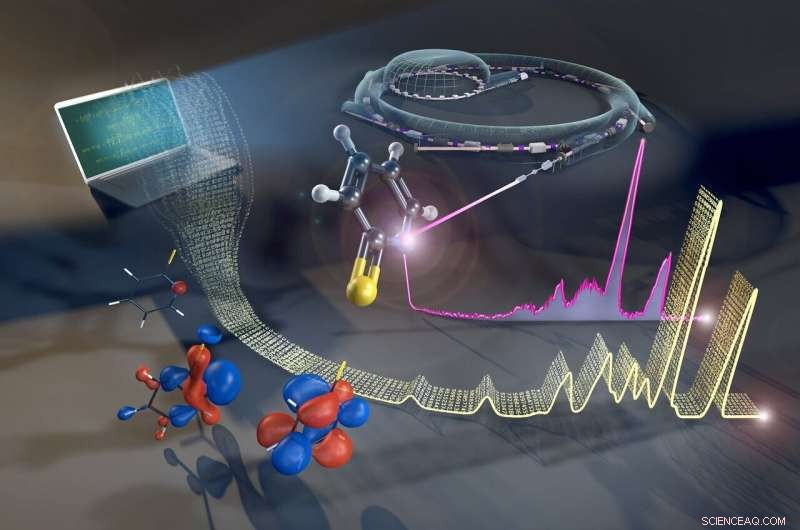
A estrutura eletrônica de moléculas complexas pode ser avaliada pelo método de espalhamento inelástico de raios-X ressonante (RIXS) em BESSY II. Crédito:Martin Künsting / HZB
Moléculas compostas por muitos átomos são estruturas complexas. Os elétrons externos são distribuídos entre os diferentes orbitais, e sua forma e ocupação determinam o comportamento químico e a reatividade da molécula. A configuração desses orbitais pode ser analisada experimentalmente. Fontes síncrotron como BESSY II fornecem um método para esse propósito:Espalhamento inelástico de raios-X ressonante (RIXS). Contudo, para obter informações sobre os orbitais a partir de dados experimentais, simulações de química quântica são necessárias. Os tempos de computação típicos para moléculas maiores levam semanas, mesmo em computadores de alto desempenho.
Agilizando a avaliação
"Até agora, esses cálculos foram realizados principalmente após as medições, “explica o químico teórico Dr. Vinicius Vaz da Cruz, pós-doutorado na equipe do Prof. Dr. Alexander Föhlisch. Juntamente com o especialista em RIXS, Dr. Sebastian Eckert, também pós-doutorado na equipe de Föhlisch, eles desenvolveram um novo procedimento sofisticado que acelera muitas vezes a avaliação.
"Com nosso método, leva alguns minutos e não precisamos de um supercomputador para isso, funciona em máquinas desktop, "diz Eckert. Os cientistas do HZB testaram o método na molécula 2-tiopiridona, um sistema modelo para transferência de prótons, que são processos essenciais em células e organismos vivos. Apesar do curto tempo de computação, os resultados são precisos o suficiente para serem muito úteis.
"Este é um grande passo em frente, "enfatiza Föhlisch." Podemos percorrer muitas opções com antecedência e conhecer a molécula, por assim dizer. Além disso, este método também torna possível simular moléculas muito mais complexas e interpretar os dados obtidos experimentalmente de uma forma significativa. "O físico experimental Eckert acrescenta:" Agora também podemos executar as simulações durante a medição e ver imediatamente onde pode ser particularmente emocionante para dar uma olhada mais de perto. "
O procedimento é uma extensão da teoria funcional da densidade dependente do tempo bem estabelecida e altamente eficiente, que é muito mais rápido do que os conceitos tradicionais para simular o processo RIXS. "A simplicidade do método permite um grande grau de automatização, "diz Vaz da Cruz:" Pode ser usado como caixa-preta. "