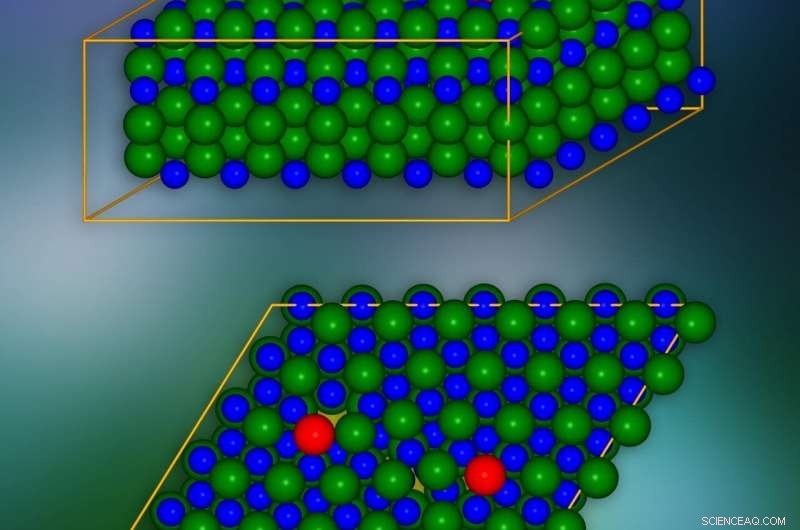
Modelo de cristal de carboneto de silício com deslocamentos de borda introduzidos em locais marcados em vermelho. Um único plano cristalográfico é apresentado na parte inferior. Os locais onde as cargas elétricas podem "vazar" para as camadas vizinhas são marcados em amarelo. Crédito:IFJ PAN
Imperfeições da estrutura cristalina, especialmente deslocamentos de borda de natureza alongada, modificar profundamente as propriedades básicas de todo o material e, em consequência, limitar drasticamente suas aplicações. Usando carboneto de silício como exemplo, físicos da Cracóvia e de Varsóvia mostraram que mesmo esses defeitos que exigem muito do computador podem ser examinados com sucesso com precisão atômica por meio de um sistema habilmente construído, pequeno em tamanho, modelo.
A matemática adora perfeição. Infelizmente, a perfeição não ama a realidade física. Os teóricos que modelam cristais há muito tentam incluir defeitos em estruturas cristalinas reais e prever seu impacto nas propriedades físicas dos materiais. Os modelos, com base nos resultados de vários experimentos, descreveram mudanças nas propriedades básicas de um material sem explicar as verdadeiras causas e efeitos dos fenômenos ocorrentes.
Um novo modelo de carboneto de silício (SiC), construído por físicos do Instituto de Física Nuclear da Academia Polonesa de Ciências (IFJ PAN) em Cracóvia, permitiu-lhes demonstrar que agora é possível estudar cristais ab initio com defeitos complexos como deslocamentos de borda e explicar suas características por processos que ocorrem em escala atômica. Este resultado espetacular, recentemente apresentado na conferência Multiscale Phenomena in Molecular Matter 2019 em Cracóvia, foi alcançado pelos físicos PAN da IFJ em cooperação com o Instituto de Pesquisa Tecnológica Fundamental da Academia Polonesa de Ciências e o Instituto de Física de Alta Pressão da Academia Polonesa de Ciências, ambos localizados em Varsóvia.
“Tentamos encontrar os mecanismos responsáveis, em nível atômico, por diminuir a tensão de ruptura nos cristais de carboneto de silício. Nossos cálculos ab initio levam a uma compreensão qualitativa do problema e contribuem para explicar os detalhes desse fenômeno, "diz o Dr. Jan Lazewski, professor do IFJ PAN.
Os cálculos ab initio têm agora uma longa história relacionada ao Prêmio Nobel para Walter Kohn e John Pople em 1998 (no entanto, para simulações de defeitos de cristal linear, eles só foram introduzidos recentemente). Este termo é usado para descrever cálculos realizados usando equações da mecânica quântica, apoiado apenas pelo conhecimento sobre a estrutura do átomo e a simetria dos cristais. Não há informações diretas de experimentos em tais modelos, o que significa que também podem ser usados para analisar materiais que nunca foram estudados ou mesmo sintetizados antes. Por causa da complicação relativamente substancial do problema, até agora os cálculos ab initio funcionaram, no máximo, no caso de defeitos pontuais, relacionadas a lacunas (átomos ausentes ou buracos na estrutura do cristal), bem como adições introduzidas no cristal.
Não foi sem razão que os pesquisadores da Cracóvia usaram carboneto de silício. As propriedades desse semicondutor são tão interessantes que no passado ele foi até considerado um sucessor do silício. Sua lacuna de banda (a barreira que a carga precisa superar para ir da banda de valência para a banda de condução e conduzir a corrente) é quase três vezes maior do que no silício, a densidade de corrente de condução permissível - duas vezes maior, a capacidade de dissipar calor - mais de três vezes maior, e a frequência de corte da operação do cristal até seis vezes maior. Além disso, sistemas de carboneto de silício podem operar em temperaturas de até 650 graus Celsius, enquanto os sistemas de silício já começam a ter problemas a 120 graus Celsius. SiC também tem um alto ponto de fusão, É difícil, resistente ao ácido e à radiação. Suas desvantagens incluem acima de tudo o preço:enquanto os wafers de silício de duas polegadas custam apenas alguns dólares, o valor de bolachas de carboneto de silício semelhantes chega a milhares. Cristais de carboneto de silício de baixa qualidade são um material abrasivo popular, também usado em coletes à prova de balas e nos discos de freio dos carros mais caros do mundo, como Lamborghini ou Bugatti. Cristais de alta qualidade são usados na produção de espelhos para telescópios e em dispositivos de alta tensão com alta resistência à temperatura.
No nível atômico, os cristais de carboneto de silício são compostos de muitas camadas planas dispostas uma em cima da outra. Cada camada se assemelha a um favo de mel:consiste em células hexagonais nas quais as moléculas de carboneto de silício estão localizadas verticalmente nos cantos. Cada duas camadas adjacentes podem ser combinadas de três maneiras. Os 'sanduíches' multicamadas com diferentes layouts criam os chamados politipos, dos quais existem mais de 250 no caso do carboneto de silício. O grupo da IFJ PAN usou o polimorfo 4H-SiC.
"Ao modelar tais estruturas, um dos principais problemas é a complexidade computacional. Um modelo de cristal puro, desprovido de misturas ou deslocamentos, é caracterizado por uma grande simetria e pode ser calculado em poucos minutos. Para realizar o cálculo de um material com deslocamento, precisamos de meses trabalhando em um computador de alta potência, "enfatiza o Dr. Pawel Jochym, professor do IFJ PAN.
Os problemas com deslocamentos de aresta resultam da escala de sua influência na estrutura cristalina do material. Como uma ilustração, eles podem ser comparados ao problema de disfarçar uma lacuna em uma fileira de ladrilhos no chão. A lacuna pode ser 'camuflada' movendo os ladrilhos das filas adjacentes, mas o defeito sempre permanecerá visível. Deslocamentos de borda resultantes da falta de comprimentos inteiros ou regiões de átomos / moléculas em camadas de cristal individuais agem de forma semelhante, afetando as posições de átomos e moléculas em muitas camadas adjacentes. E uma vez que os deslocamentos podem se estender por longas distâncias, na prática, os distúrbios causados por eles incluem todo o cristal.
Os fenômenos mais interessantes ocorrem no núcleo de deslocamento, ou seja, na vizinhança da borda da camada danificada da rede de cristal. A fim de eliminar os efeitos de longo alcance causados por um único deslocamento, e, assim, reduzir significativamente o número de átomos em consideração, um truque foi empregado:um segundo deslocamento do efeito oposto foi introduzido. Desta maneira, o impacto do primeiro deslocamento em distâncias mais longas foi compensado.
O modelo de cristal de SiC consistia em cerca de 400 átomos. As simulações mostraram que nas camadas de cristais, ao longo da borda do núcleo do defeito, 'túneis' aparecem na forma de canais com densidade de carga reduzida. Eles reduzem a barreira de potencial localmente e fazem com que as cargas elétricas "vazem" da banda de valência. Além disso, na lacuna proibida, que no isolador garante uma falta de condutividade elétrica, aparecem condições que reduzem sua largura e eficácia em limitar o fluxo de carga. Foi mostrado que esses estados se originam de átomos localizados no núcleo de deslocamento.
"A situação pode ser comparada a uma profunda, ravina íngreme que um esquilo está tentando atravessar. Se o fundo da ravina estiver vazio, o esquilo não chegará ao outro lado. Contudo, se houver várias árvores no fundo que são altas o suficiente, o esquilo pode pular para o outro lado da ravina. No cristal que modelamos, os esquilos são as cargas elétricas, a banda de valência é uma borda da ravina, a banda de condução é a outra, e as árvores são os estados mencionados anteriormente associados aos átomos do núcleo de deslocamento, "diz o Prof. Lazewski.
Agora que os mecanismos responsáveis por diminuir o limiar da barreira de energia tornaram-se conhecidos em nível atômico, há um grande espaço para experimentação. O mecanismo proposto deverá ser verificado para poder utilizá-lo para limitar a influência negativa dos defeitos testados. Felizmente, já existem possibilidades técnicas para isso.
“O futuro vai verificar se nossas ideias serão confirmadas na íntegra. estamos confiantes sobre o destino de nosso modelo e a abordagem apresentada para simular deslocamentos de borda. Já sabemos que o modelo ab initio provou seu valor em confronto com certos dados experimentais, "conclui o Prof. Jochym.