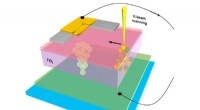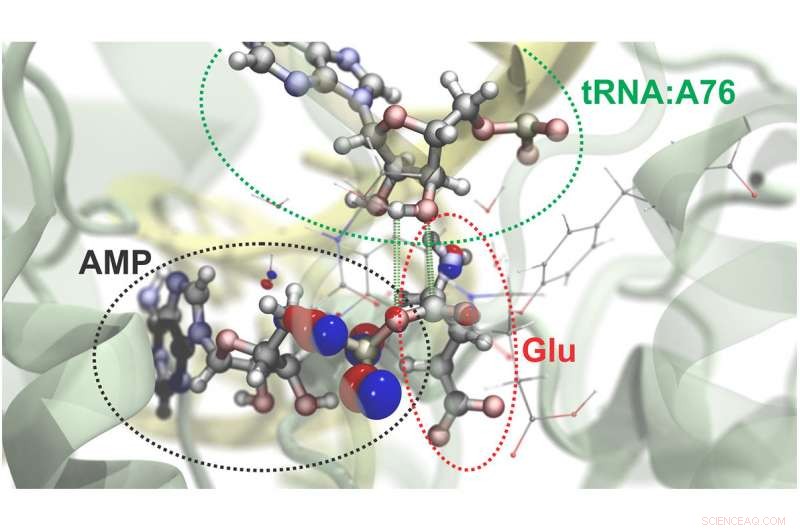
Os pesquisadores podem simular a dinâmica atômica e subatômica em grandes sistemas moleculares. Aqui está uma visualização do processo pelo qual o aminoácido glutamato (Glu) é anexado a uma região específica de seu RNA de transferência (tRNA). Uma molécula rica em energia, ATP, conduz essa reação e é convertido em AMP no processo. As bolhas vermelhas e azuis representam a probabilidade de encontrar elétrons em regiões específicas. As linhas pontilhadas verdes delineiam os átomos que se ligam nesta reação química. Crédito:Rafael Bernardi, Zan Luthey-Schulten e Marcelo Melo
Os cientistas construíram um "microscópio computacional" que pode simular as forças atômicas e subatômicas que conduzem as interações moleculares. Esta ferramenta irá agilizar os esforços para compreender a química da vida, modelar grandes sistemas moleculares e desenvolver novos agentes farmacêuticos e industriais, dizem os pesquisadores.
Eles relatam suas descobertas no jornal Métodos da Natureza .
Os cientistas combinaram duas abordagens computacionais usadas para simular interações moleculares. O primeiro, um programa de dinâmica molecular em nanoescala conhecido como NAMD, usa métodos da mecânica clássica para modelar a estrutura e simular o comportamento de centenas de milhões de átomos individuais. O segundo programa se aproxima do reino subatômico, simulando as interações de prótons, nêutrons e elétrons. A modelagem nesta escala de mecânica quântica exige muito poder computacional, então os pesquisadores implementaram um método para particionar grandes moléculas em regiões de mecânica clássica e quântica. Isso permite que eles concentrem seus recursos computacionais em pequenas regiões envolvidas em interações críticas, como a formação ou quebra de ligações químicas.
Programas de mecânica molecular e mecânica quântica estão disponíveis há anos, e outras equipes trabalharam para combiná-los, disse o professor de química da Universidade de Illinois, Zaida (Zan) Luthey-Schulten, que liderou a nova pesquisa com seu marido, U. of I. professor de física Klaus Schulten. Mas o novo esforço simplifica o processo de configuração, realizar e analisar as simulações.
“Nós o configuramos para que os pesquisadores possam escolher facilmente como irão particionar seus próprios sistemas, "Luthey-Schulten disse." Meus próprios alunos estão experimentando, e a maioria deles é capaz de fazer isso sem muita dificuldade. "
Schulten desenvolveu o NAMD em Illinois em 1995, combinando-o com um software de visualização, VMD, que permite aos pesquisadores observar o desdobramento das interações moleculares em grande escala. Schulten, que morreu em 2016, equiparou esta abordagem a "construir um microscópio computacional."
O microscópio computacional é ideal para modelar traços estruturais e movimentos de grandes complexos. Por exemplo, em 2013, Schulten e seus colegas usaram o NAMD para modelar o capsídeo do HIV, que é composto por mais de 1, 300 proteínas idênticas que se montam em uma estrutura semelhante a uma gaiola que protege o vírus até que ele entre na célula hospedeira. Essa simulação foi responsável pelas interações de mais de 64 milhões de átomos e exigiu o uso do supercomputador Blue Waters no National Center for Supercomputing Applications na U. of I. O novo estudo também fez uso do Blue Waters, desta vez para melhorar a resolução do microscópio computacional.

Da esquerda, aluno de graduação Marcelo Melo, professora de química Zaida Luthey-Schulten, o pesquisador de pós-doutorado Rafael Bernardi e seus colegas desenvolveram uma nova abordagem para modelar grandes interações moleculares em escalas atômicas e subatômicas. Seu trabalho agiliza o método para outros cientistas e alunos. Crédito:L. Brian Stauffer
O software NAMD é projetado para descrever o comportamento de átomos individuais. Mas átomos individuais envolvidos em reações e interações químicas específicas nem sempre se comportam como suas contrapartes em outros lugares. Para entender como eles variam, é necessário examinar mais de perto as forças subatômicas em jogo. Isso é particularmente importante nas regiões dinâmicas das moléculas - por exemplo, aqueles lugares onde as ligações químicas são feitas ou quebradas, disseram os pesquisadores.
No novo estudo, a equipe de pesquisa em Illinois uniu-se aos especialistas em QM Frank Neese, do Instituto Max Planck para Pesquisa de Carvão em Mulheim an der Ruhr, Alemanha; e Gerd B. Rocha, da Universidade Federal da Paraíba, em João Pessoa, Brasil.
Como demonstração da nova abordagem, os pesquisadores simularam o comportamento químico de RNAs de transferência, moléculas que desempenham um papel fundamental na tradução da informação genética em proteínas. Usando NAMD, eles modelaram a estrutura molecular geral do tRNA no momento em que uma proteína especial carrega um aminoácido no tRNA. Eles dividiram dois locais do complexo em regiões que exigiam a abordagem da mecânica quântica mais focada. (Assista a um filme da simulação.)
As simulações subatômicas das interações das duas regiões permitiram que a equipe fizesse simulações de quatro cenários diferentes que permitiriam ao tRNA funcionar da mesma forma que na célula. Suas simulações revelaram que uma das quatro vias químicas potenciais era mais favorável do ponto de vista energético do que as outras e, portanto, mais provável de ocorrer.
Os pesquisadores também usaram vários métodos para particionar o complexo tRNA entre as regiões MM e QM e relataram cada abordagem.
"Não escolhemos apenas um caminho; escolhemos tantos quanto possível. Damos liberdade ao usuário. A forma como você o estrutura realmente depende do sistema específico que você está estudando, "disse U. de I. pesquisador de pós-doutorado Rafael Bernardi, co-autor do estudo com o pós-graduando Marcelo Melo.
"Não fazemos todo o sistema quântico mecanicamente porque levaria uma eternidade para calcular, "Disse Melo.
"O NAMD foi projetado - e essa era a visão do meu marido - para tratar sistemas realmente grandes, "Luthey-Schulten disse." Agora podemos adicionar a escala subatômica a isso, abrindo novas possibilidades de pesquisa. "