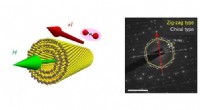
Modelo estrutural da fibrila peptídica Alzheimer Amyloid A-beta 1-42 derivada de uma estrutura experimental (PDB:2MXU). Os agregados fibrilares atuam como toxinas celulares no início e na progressão da doença de Alzheimer. Crédito:Emanuel Peter
Proteínas, os onipresentes burros de carga da bioquímica, são moléculas enormes cuja função depende de como elas se dobram em estruturas intrincadas. Para entender como essas moléculas funcionam, pesquisadores usam modelagem de computador para calcular como as proteínas se dobram.
Agora, um novo algoritmo pode acelerar essas simulações vitais, permitindo-lhes modelar fenômenos que antes estavam fora de alcance. Os resultados podem eventualmente ajudar os cientistas a entender e tratar melhor doenças como Alzheimer, disse Emanuel Peter, um químico da Universidade de Regensburg. Seu trabalho na nova técnica é descrito esta semana em The Journal of Chemical Physics .
Simulações convencionais, usando dinâmica molecular e métodos de Monte Carlo, têm sido bem-sucedidos em geral na modelagem de moléculas biológicas como proteínas. Para determinar como as proteínas se dobram, a simulação busca configurações que correspondam a estados de energia cada vez mais baixos. Eventualmente, encontra o estado de energia mais baixo, o que dá uma dobra estável. Mas conforme a simulação procura, pode encontrar uma configuração com uma energia ligeiramente superior, que forma uma barreira que impede o algoritmo.
Como resultado dessas desacelerações, os métodos convencionais só podem simular comportamentos moleculares que ocorrem em escalas de tempo curtas de algumas centenas de microssegundos. Muitos fenômenos, como certas dobras de proteína ou uma ligação de droga a um alvo potencial, acontecer ao longo de alguns segundos, minutos ou mesmo dias. Simular escalas de tempo tão longas exigiria muito tempo de computação apenas com as abordagens convencionais.
Para acelerar as simulações, pesquisadores podem injetar energia no sistema, que empurra o modelo sobre quaisquer barreiras de energia. Mas um dos maiores desafios para esses métodos é definir as coordenadas que descrevem o sistema, que, por exemplo, pode ser o comprimento entre os átomos na molécula, e os ângulos entre as ligações. Tradicionalmente, os pesquisadores definem as coordenadas antes de iniciar a simulação. Cada passo ao longo de cada coordenada depende do passo anterior. Mas essa dependência pode influenciar a simulação.
O novo algoritmo de Peter evita esse viés. Ele encontrou um sistema de coordenadas generalizado no qual cada etapa de tempo não depende da etapa anterior. "Apenas alguns parâmetros são necessários, e nenhuma intuição humana é necessária, o que pode potencialmente influenciar o resultado da simulação, " ele disse.
Para testar o novo algoritmo, Peter usou para modelar água, um peptídeo chamado dialanina, o dobramento de outro peptídeo chamado TrpCage, e a aglomeração de beta-amilóide 25-35, que são fragmentos de proteínas associados à doença de Alzheimer. Em cada caso, sua técnica relata ter acelerado as simulações. E as simulações de beta-amiloide podem ajudar a explicar por que o Alzheimer é difícil de tratar.
Na doença de Alzheimer, fragmentos de proteína beta-amilóide se agregam, formando uma placa dura que se acumula entre os neurônios e os interrompe. O beta-amilóide também é uma toxina, levando à morte de células neuronais e degeneração da função neuronal. As novas simulações sugerem que a beta-amiloide pode assumir uma variedade de estruturas. Essa flexibilidade estrutural pode ser o motivo pelo qual algumas drogas que tentam inibir a agregação não tiveram sucesso, Disse Peter. Quando essas drogas se ligam ao beta-amilóide, o beta-amilóide apenas muda de forma, permitindo que eles continuem se agrupando. A droga é incorporada ao agregado e à placa.
Este tipo de flexibilidade estrutural, chamada entropia de conformação, também é uma característica fundamental em outros peptídeos que formam placas tóxicas em doenças como a doença de Huntington, Diabetes tipo 2, e doença de Parkinson. O novo algoritmo pode, portanto, ser útil para a compreensão dessas outras doenças também.