Os cientistas da IBM desenvolveram uma nova abordagem para simular moléculas em um computador quântico que pode um dia ajudar a revolucionar a química e a ciência dos materiais. Os cientistas usaram com sucesso seis qubits em um processador quântico de sete qubits desenvolvido especificamente para resolver o problema da estrutura molecular do hidreto de berílio (BeH2) - a maior molécula simulada em um computador quântico até hoje. Os resultados demonstram um caminho de exploração para sistemas quânticos de curto prazo para aprimorar nossa compreensão de reações químicas complexas que podem levar a aplicações práticas. Crédito:Kandala et al .; Natureza
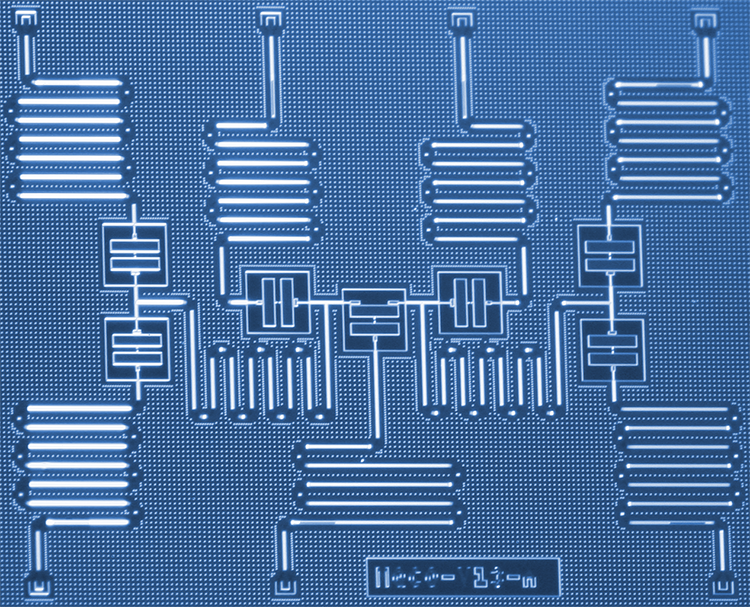
Simular moléculas em computadores quânticos ficou muito mais fácil com o hardware quântico supercondutor da IBM. Em um artigo de pesquisa recente publicado em Natureza , Variacional Quantum Eigensolver com hardware eficiente para pequenas moléculas e ímãs quânticos, implementamos um novo algoritmo quântico capaz de computar com eficiência o estado de menor energia de pequenas moléculas. Ao mapear a estrutura eletrônica dos orbitais moleculares em um subconjunto de nosso processador quântico de sete qubit construído especificamente, estudamos moléculas até então inexploradas com computadores quânticos, incluindo hidreto de lítio (LiH) e hidreto de berílio (BeH2). A codificação particular de orbitais para qubits estudados neste trabalho pode ser usada para simplificar simulações de moléculas ainda maiores e esperamos a oportunidade de explorar tais simulações maiores no futuro, quando o poder computacional quântico (ou "volume quântico") dos sistemas IBM Q aumentou.
Embora BeH2 seja a maior molécula já simulada por um computador quântico até hoje, o modelo considerado da própria molécula ainda é simples o suficiente para que os computadores clássicos simulem com exatidão. Isso tornou um caso de teste para empurrar os limites do que nosso processador de sete qubit poderia alcançar, aprofundar nossa compreensão dos requisitos para aumentar a precisão de nossas simulações quânticas, e estabelecer os elementos básicos necessários para explorar esses estudos de energia molecular.
As melhores simulações de moléculas hoje são executadas em computadores clássicos que usam métodos aproximados complexos para estimar a energia mais baixa de um hamiltoniano molecular. Um "hamiltoniano" é um operador de energia mecânica quântica que descreve as interações entre todos os orbitais de elétrons e núcleos dos átomos constituintes. O estado de "energia mais baixa" do hamiltoniano molecular dita a estrutura da molécula e como ela irá interagir com outras moléculas. Essas informações são críticas para os químicos projetarem novas moléculas, reações, e processos químicos para aplicações industriais.
Qubit:Orbital
Embora nosso processador quântico de sete qubit não seja totalmente corrigido e tolerante a falhas, os tempos de coerência dos qubits individuais duram cerca de 50 µs. Portanto, é realmente importante usar um algoritmo quântico muito eficiente para tirar o máximo proveito de nossa preciosa coerência quântica e tentar entender as estruturas moleculares. O algoritmo deve ser eficiente em termos de número de qubits usados e número de operações quânticas realizadas.

Aplicação à química quântica. a – c, Resultados experimentais (círculos pretos), superfícies de energia exata (linhas pontilhadas) e gráficos de densidade (sombreamento; ver escalas de cores) de resultados de simulações numéricas, para várias distâncias interatômicas para H2 (a), LiH (b) e BeH2 (c). Os resultados experimentais e numéricos apresentados são para circuitos de profundidade d =1. As barras de erro nos dados experimentais são menores que o tamanho dos marcadores. Os gráficos de densidade são obtidos a partir de 100 resultados numéricos em cada distância interatômica. As inserções superiores em cada painel destacam os qubits usados para o experimento e as portas de ressonância cruzada (setas, rotulado CRc – t; onde 'c' denota o qubit de controle e 't' o qubit alvo) que constituem o UENT. As inserções inferiores são representações da geometria molecular (não em escala). Para todas as três moléculas, o desvio dos resultados experimentais das curvas exatas é bem explicado pelas simulações estocásticas. Crédito:Kandala et al .; Natureza
Nosso esquema contrasta com algoritmos de simulação quântica previamente estudados, que se concentram na adaptação de esquemas clássicos de simulação molecular ao hardware quântico - e, ao fazê-lo, não levam em consideração as despesas gerais limitadas dos atuais dispositivos quânticos realistas.
Então, em vez de forçar métodos de computação clássicos para o hardware quântico, invertemos a abordagem e perguntamos:como podemos extrair a potência computacional quântica máxima de nosso processador de sete qubit?
Nossa resposta para isso combina uma série de técnicas eficientes de hardware para atacar o problema:
Com futuros processadores quânticos, que terá mais volume quântico, seremos capazes de explorar o poder desta abordagem para simulação quântica para moléculas cada vez mais complexas que estão além das capacidades clássicas de computação. A capacidade de simular reações químicas com precisão, conduz aos esforços de descoberta de novos medicamentos, fertilizantes, até mesmo novas fontes de energia sustentável.
Os experimentos que detalhamos em nosso artigo não foram executados em nossos processadores de cinco qubit e 16 qubit atualmente disponíveis publicamente na nuvem. Mas os desenvolvedores e usuários da experiência IBM Q agora podem acessar notebooks Jupyter de química quântica no repositório github QISKit. No sistema de cinco qubit, os usuários podem explorar a simulação de energia do estado fundamental para as pequenas moléculas de hidrogênio e LiH. Notebooks para moléculas maiores estão disponíveis para aqueles com acesso beta ao processador de 16 qubit atualizado.