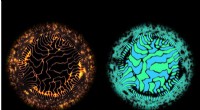
Uma representação de fatias bidimensionais aleatórias de uma função de 12 dimensões para determinar as correções de energia e frequência de uma molécula de formaldeído. Crédito:Sandia National Laboratories
Pesquisadores do Sandia National Laboratories desenvolveram novas técnicas matemáticas para avançar no estudo de moléculas no nível quântico.
Desenvolvimentos matemáticos e algorítmicos ao longo dessas linhas são necessários para permitir o estudo detalhado de moléculas de hidrocarbonetos complexos que são relevantes na combustão de motores.
Os métodos existentes para aproximar funções de energia potencial na escala quântica precisam de muito poder de computador e, portanto, são limitados a moléculas pequenas. Os pesquisadores do Sandia dizem que sua técnica vai acelerar os cálculos da mecânica quântica e melhorar as previsões feitas por modelos teóricos da química. Dada a aceleração computacional, esses métodos podem ser potencialmente aplicados a moléculas maiores.
O pesquisador de pós-doutorado em Sandia, Prashant Rai, trabalhou com os pesquisadores Khachik Sargsyan e Habib Najm no Centro de Pesquisa em Combustão de Sandia e colaborou com os químicos quânticos So Hirata e Matthew Hermes na Universidade de Illinois em Urbana-Champaign. Computando a energia em menos arranjos geométricos do que o normalmente necessário, a equipe desenvolveu métodos computacionalmente eficientes para aproximar as superfícies de energia potencial.
Uma compreensão precisa das superfícies de energia potencial, elementos-chave em praticamente todos os cálculos da dinâmica quântica, é necessário para estimar com precisão a energia e a frequência dos modos vibracionais das moléculas.
"Se pudermos encontrar a energia da molécula para todas as configurações possíveis, podemos determinar informações importantes, tais como estados estáveis de estrutura de transição molecular ou estados intermediários de moléculas em reações químicas, "Rai disse.
Os resultados iniciais desta pesquisa foram publicados em Física Molecular em um artigo intitulado "Decomposição de tensor canônico de baixa classificação de superfícies de energia potencial:aplicação à teoria da função de Green vibracional diagramática baseada em grade."

Prashant Rai, pesquisadores do Sandia National Laboratories, deixou, Habib Najm, Centro, e Khachik Sargsyan discutem técnicas matemáticas usadas para estudar o comportamento de grandes moléculas em escala quântica. Crédito:Dino Vournas
"Aproximar as superfícies de energia potencial de moléculas maiores é uma tarefa extremamente desafiadora devido ao aumento exponencial nas informações necessárias para descrevê-las com cada átomo adicional no sistema, "Rai disse." Em matemática, é denominado a Maldição da Dimensionalidade. "
Vencendo a maldição
A chave para vencer a maldição da dimensionalidade é explorar as características da estrutura específica das superfícies de energia potencial. Rai disse que essa informação de estrutura pode então ser usada para aproximar as funções dimensionais altas necessárias.
"Aproveitamos o fato de que, embora as superfícies de energia potencial possam ser altamente dimensionais, eles podem ser bem aproximados como uma pequena soma de produtos de funções unidimensionais. Isso é conhecido como estrutura de baixo escalão, onde a classificação da superfície de energia potencial é o número de termos na soma, "Rai disse." Tal suposição sobre a estrutura é bastante geral e também foi usada em problemas semelhantes em outros campos. Matematicamente, a intuição das técnicas de aproximação de classificação baixa vem da álgebra multilinear, onde a função é interpretada como um tensor e é decomposta usando técnicas de decomposição de tensor padrão. "
As correções de energia e frequência são formuladas como integrais dessas funções de energia de alta dimensão. A aproximação em um formato de baixa classificação torna essas funções facilmente integráveis, pois quebra o problema de integração para a soma dos produtos de integrais unidimensionais ou bidimensionais, portanto, os métodos de integração padrão se aplicam.
A equipe testou seus métodos computacionais em pequenas moléculas como água e formaldeído. Comparado com o método clássico de Monte Carlo, o burro de carga padrão baseado em aleatoriedade para problemas de integração de alta dimensão, sua abordagem previa energia e frequência da molécula de água que eram mais precisas, e era pelo menos 1, 000 vezes mais eficiente computacionalmente.
Rai disse que o próximo passo é aprimorar ainda mais a técnica, desafiando-a com moléculas maiores, como o benzeno.
"Estudos interdisciplinares, como química quântica e engenharia de combustão, oferecem oportunidades para a polinização cruzada de ideias, proporcionando assim uma nova perspectiva sobre os problemas e suas possíveis soluções, "Rai disse." É também um passo em direção ao uso de avanços recentes na ciência de dados como um pilar da descoberta científica no futuro. "